 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rosella Visintin | + 2002 word(s) | 2002 | 2020-08-10 08:16:21 | | | |
| 2 | Rita Xu | -547 word(s) | 1455 | 2020-09-04 04:00:18 | | |
Video Upload Options
Anaphase bridges are DNA threads stretching between the two DNA masses as cells attempt to segregate them during anaphase. Anaphase bridges arise from unresolved DNA intertwines between sister chromatids. Sister chromatid intertwines (SCIs) naturally arise during DNA replication and represent a non-proteinaceous source of cohesion between sister chromatids. SCIs and are mainly resolved in S phase, although some do persist and must be fully removed during mitosis to allow faithful chromosome segregation and avoid the arising of DNA lesions and genome instability, a hallmark of cancer development. As complete resolution of SCIs only occurs during chromosome segregation, it is not clear whether intertwines that persist in mitosis are simply an unwanted leftover or whether they have a functional role.
1. Structure and Origin of Anaphase Bridges
Anaphase bridges originate from unresolved SCIs. DNA intertwines are usually classified based on their molecular structure, which reflects their origin (Figure 1). Three types of intertwines are recognized, namely short regions of un-replicated DNA, recombination intermediates and DNA catenanes. Although certain DNA linkages are more commonly retained at specific loci, since different types of linkages can form anaphase bridges at the same region, their position in the genome is not sufficient to infer their molecular structure.
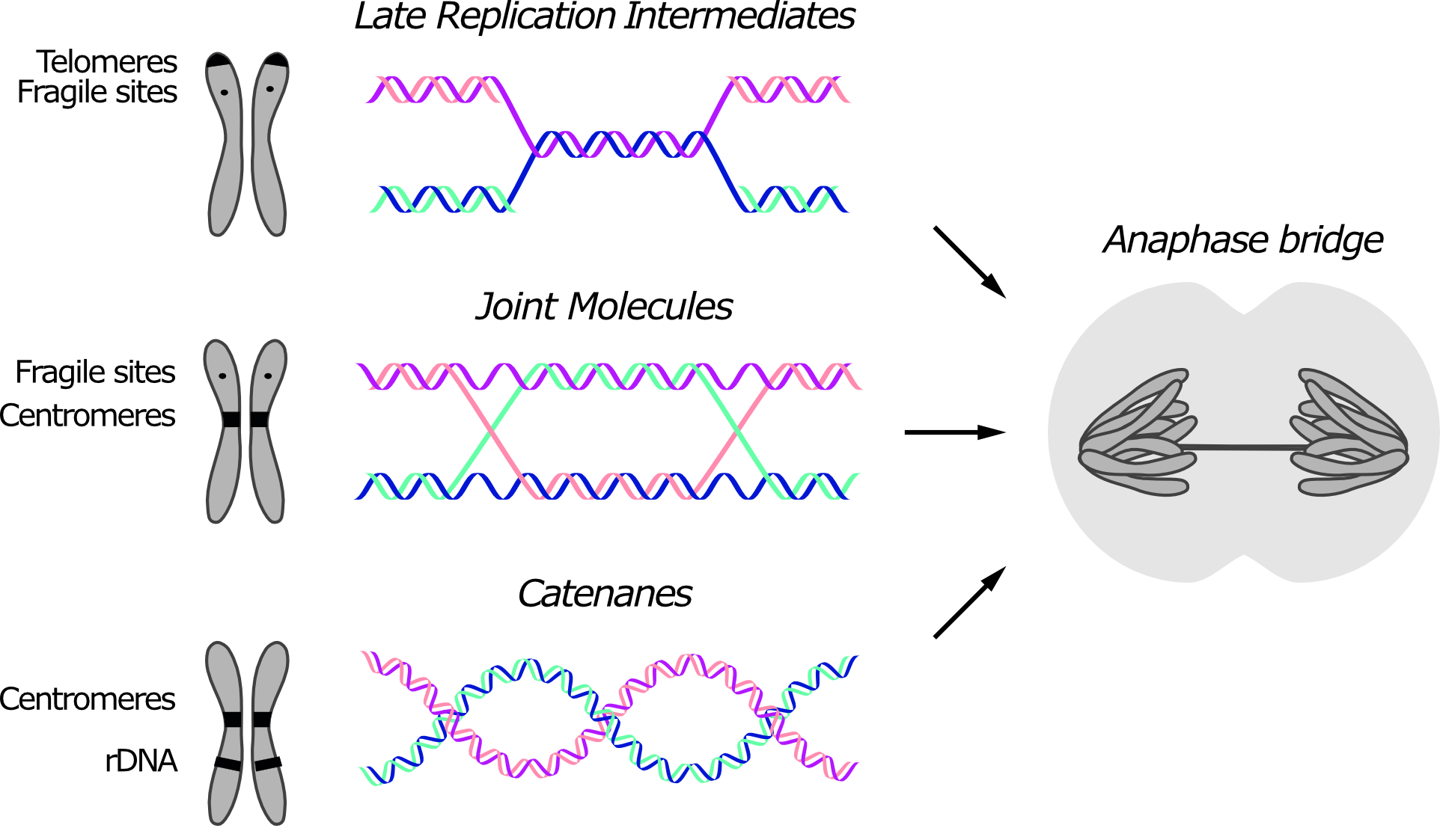
Figure 1. Three types of sister chromatid intertwines (SCIs) are observed during mitosis. Molecular structures of late replication intermediates, joint molecules (here exemplified by a double Holliday junction) and catenanes are shown. Genomic loci at which each type of SCI was observed are indicated in the schematic chromosome. The consequence of persistent SCIs in mitosis – a.k.a. anaphase bridges – is shown in the drawn cell.
Although the number of anaphase bridges increases in the presence of stress, such as treatment with replication inhibitors or impairment of DNA repair pathways, DNA bridges are observed even in the absence of stressors and in untransformed cells, especially at the centromeres in mammalian cells [1][2]. In addition, anaphase bridges also form when cells attempt to segregate dicentric chromosomes that result from telomere-to-telomere fusion [3][4][5].
Anaphase bridges can be classified as either chromatin bridges or ultra-fine bridges (UFBs), based on whether or not the DNA is chromatinized (i.e., packaged with histones and other proteins) and can be stained with DNA dyes like Hoechst. In unperturbed conditions, chromatin bridges are rare and, in yeast, they are mostly attributed to unresolved recombination intermediates [6]. On the other hand, UFBs can be observed in the majority of cells in anaphase, and they are enriched at the centromere in human cells [1][6]. The same kind of stressor can often induce formation of both types of bridges (i.e., chromatinized and non-chromatinized), suggesting that both can arise from the same kind of DNA intertwining. Currently, it is not clear what determines the chromatinized status of DNA bridges. Chromatin bridges may evolve into UFBs, according to the stage of their resolution. Alternatively, the chromatinized status of a bridge may be determined at the time of its formation. In the latter case, DNA chromatinization may depend on several factors, such as the extent of intertwining or the timing of recognition during mitosis.
2. A Time to Bridge and a Time to Rupture
Persistent anaphase bridges, independently of the source, can cause gross chromosomal rearrangements, a hallmark of cancer development. Chromosomal rearrangements are likely a consequence of DSBs that are formed when the bridge eventually breaks. Indeed, it was recently shown that even one single unresolved bridge triggers a cascade of events that result in increasing amounts of genome rearrangements [5]. Highly-proliferating cancer cells are known to be under increased replicative stress [7] and, since replicative stress enhances anaphase bridges [2][6][8], it is reasonable to think that these structures may arise in cancer cells with a higher frequency compared to normal conditions. If this scenario was true, anaphase bridges, being both a consequence of replicative stress and a cause of genome instability, could be considered a driver of tumor progression.
Several models have been proposed to explain how anaphase bridges drive genomic instability. The first model is the so-called breakage-fusion-bridge (BFB) cycle, described by Barbara McClintock [9]. According to this idea, DNA bridges that are cleaved during cytokinesis are re-sealed in the daughter cells with other chromosome pieces, causing gross genome rearrangements in the form of reciprocal exchanges of chromosome arms. The BFB model was recently extended by Pellman and colleagues. In particular, they found that bridge rupture, in addition to causing the chromosomal aberrations predicted by the BFB model, also leads to more complex rearrangements, namely chromothripsis [5]. Chromothripsis is the shattering of one or a few chromosomes, followed by stitching of the fragments in a random order. Chromothripsis is commonly observed starting from the second generation after bridge occurrence, and it is caused by abnormal replication of the bridging DNA during mitosis [5]. In addition to BFB, Chan and colleagues proposed an alternative model for the rupture of UFBs arising from recombination intermediates, called sister-chromatid rupture and bridging [10]. In this case, in contrast to BFB, the cleavage occurs shortly after anaphase onset and is independent of cytokinesis. According to this model, the intertwined sister chromatids break along the chromosome axis at the site where the bridge originates, while the bridge itself remains intact. As a result, the two sister arms are fused through the bridge and are thereby inherited by one of the two daughter cells. This mechanism is predicted to lead to specific chromosome rearrangements, including whole-arm deletions and translocations.
How do anaphase bridges break? It seems reasonable to think that threads of uncompacted – or even unchromatinized and single-stranded – DNA are fragile and can easily be broken by spindle elongation or by the actomyosin ring during cytokinesis. Although the pulling force generated by the spindle is estimated to be far too weak to break mitotic chromosomes [11], it could be sufficient to break a single DNA thread [12]. Therefore, spindle elongation may be able to break anaphase bridges, especially if they contain ssDNA segments, such as the ones generated by the processing of JMs and LRIs. Nevertheless, strong evidence of DNA linkages broken in this fashion in vivo is currently lacking. Instead, there is evidence for cytokinesis being able to sever anaphase bridges. For example cytokinesis was proved to be responsible for the breakage of chromatin bridges in Top2 and condensin yeast mutants [13][14].
In order to prevent the severe DNA damage caused by rupture of DNA threads during cell abscission, cells have evolved a mechanism that delays cytokinesis in the presence of chromatin trapped on the site of cleavage. In yeast cells, this mechanism is termed the NoCut checkpoint, and it requires the activity of Aurora B/Ipl1 at the spindle midzone [15][16]. The NoCut is sensitive to SCIs coming from various sources, including replicative stress and lack of condensin or Top2 activity, but it fails to detect bridging from dicentric chromosomes [17]. Aurora B acts as a sensor for chromatin at the cleavage plane and, in the presence of DNA bridges, inhibits completion of abscission and stabilizes the spindle [15][16][17]. The activation of the NoCut checkpoint could provide additional time for the removal of the bridges in anaphase, although cells eventually divide if the bridge cannot be resolved.
In yeast, cytokinesis always leads to rupture of the anaphase bridges, likely because the yeast cell wall represents an impenetrable barrier for DNA filaments. Conversely, in mammalian cells, cytokinesis in the presence of unresolved bridges can have different outcomes, all of which can jeopardize genomic stability. First, as already mentioned, bridges can be cleaved during abscission, causing DSBs [3]. The second option is furrow regression [18]. In this case, although ingression of the cleavage furrow is initiated, cell division is never completed and results in tetraploidy, a condition that can cause senescence/apoptosis or, in some cases, initiate genomic instability. Thirdly, a mechanism reminiscent of the yeast NoCut checkpoint, called abscission checkpoint, can come into play (reviewed in [19]). Similar to the NoCut, the abscission checkpoint relies on Aurora B at the cleavage site, acting as a sensor of chromatin and delaying abscission in the presence of DNA bridges. Activation of this checkpoint triggers abscission failure and bridge stabilization, thus preventing furrow regression and tetraploidization. In the case of abscission failure, after cytokinesis the daughter cells remain linked by the intact DNA thread, but behave like separate identities [18]. Indeed, in human cells chromatin bridges rarely break during anaphase spindle elongation and they are often retained until the next G1 [3][18][20]. However, even in this case, the bridge eventually breaks and, while the nuclease TREX1 was suggested to participate in this process [3], the main factor determining the rupture of the bridge seems to be the mechanical stretching imposed by the daughter cells migrating away from each other [5]. Irrespective of the mechanism, once again the rupture of the bridge promotes chromosomal rearrangements and chromothripsis [3][5].
References
- Chan, K.-L.; North, P.S.; Hickson, I.D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007, 26, 3397–3409, doi:10.1038/sj.emboj.7601777.
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760, doi:10.1038/ncb1882.
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654, doi:10.1016/j.cell.2015.11.054.
- Nera, B.; Huang, H.-S.; Lai, T.; Xu, L. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat. Commun. 2015, 6, 10132, doi:10.1038/ncomms10132.
- Umbreit, N.T.; Zhang, C.-Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368, doi:10.1126/science.aba0712.
- Doksani, Y. The Response to DNA Damage at Telomeric Repeats and Its Consequences for Telomere Function. Genes 2019, 10, doi:10.3390/genes10040318.
- Germann, S.M.; Schramke, V.; Pedersen, R.T.; Gallina, I.; Eckert-Boulet, N.; Oestergaard, V.H.; Lisby, M. TopBP1/Dpb11 binds DNA anaphase bridges to prevent genome instability. J. Cell Biol. 2014, 204, 45–59, doi:10.1083/jcb.201305157.
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253, doi:10.1038/ncb2201.
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123, doi:10.1038/nature09745.
- Barefield, C.; Karlseder, J. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res. 2012, 40, 7358–7367, doi:10.1093/nar/gks407.
- Torres-Rosell, J.; De Piccoli, G.; Cordon-Preciado, V.; Farmer, S.; Jarmuz, A.; Machin, F.; Pasero, P.; Lisby, M.; Haber, J.E.; Aragón, L. Anaphase onset before complete DNA replication with intact checkpoint responses. Science 2007, 315, 1411–1415, doi:10.1126/science.1134025.
- Ivanova, T.; Maier, M.; Missarova, A.; Ziegler-Birling, C.; Dam, M.; Gomar-Alba, M.; Carey, L.B.; Mendoza, M. Budding yeast complete DNA synthesis after chromosome segregation begins. Nat. Commun. 2020, 11, 2267, doi:10.1038/s41467-020-16100-3.
- Le Beau, M.M.; Rassool, F.V.; Neilly, M.E.; Espinosa, R.; Glover, T.W.; Smith, D.I.; McKeithan, T.W. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: Implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998, 7, 755–761, doi:10.1093/hmg/7.4.755.
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290, doi:10.1038/nature16139.
- Pedersen, R.T.; Kruse, T.; Nilsson, J.; Oestergaard, V.H.; Lisby, M. TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J. Cell Biol. 2015, 210, 565–582, doi:10.1083/jcb.201502107.
- Prado, F. Homologous Recombination: To Fork and Beyond. Genes 2018, 9, doi:10.3390/genes9120603.
- Liberi, G.; Maffioletti, G.; Lucca, C.; Chiolo, I.; Baryshnikova, A.; Cotta-Ramusino, C.; Lopes, M.; Pellicioli, A.; Haber, J.E.; Foiani, M. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005, 19, 339–350, doi:10.1101/gad.322605.
- Branzei, D.; Sollier, J.; Liberi, G.; Zhao, X.; Maeda, D.; Seki, M.; Enomoto, T.; Ohta, K.; Foiani, M. Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 2006, 127, 509–522, doi:10.1016/j.cell.2006.08.050.
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation regulates Rad18-mediated template switch. Nature 2008, 456, 915–920, doi:10.1038/nature07587.
- Chan, Y.W.; Fugger, K.; West, S.C. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat. Cell Biol. 2018, 20, 92–103, doi:10.1038/s41556-017-0011-1.



