1000/1000
Hot
Most Recent
 +1 point
+1 point
The prion organotypic slice culture assay (POSCA) is a cerebellar slice culture that was originally developed to take advantage of the transmissible nature of prions, propagating prion infection ex vivo. Because much of the cytoarchitexture is preserved, this system allows the study of pathogenesis in an open system and is amenable to manipulation of cell types and testing of therapeutics. The culture has been adapted to other brain areas, different prion strains, and has also been applied to other neurodegenerative diseases that are prion-like in their transmissible propagation of protein misfolding, including Alzheimer’s disease, frontotemporal dementia, Parkinson’s disease, amyotrophic lateral sclerosis and Huntington’s disease. This entry provides a review of POSCA as used in the prion field. For a review of its application to other neurodegenerative diseases, please see the associated review article in Biomolecules.
Prions are infectious proteins that cause rapidly progressive, fatal neurodegenerative disorders [1]. Such diseases include Creutzfeldt Jakob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) in cows, scrapie in sheep, and chronic wasting disease (CWD) in cervids [2]. Prion diseases are caused by the misfolding of the prion protein (PrP) from its non-toxic cellular state (PrPC) to PrPSc, which exists as soluble oligomers and beta-sheet-rich fibrils and aggregates. Because PrPSc self-templates the misfolding of PrPC, PrPSc is infectious and spreads from cell-to-cell and host-to-host [1][2]. In humans, prion disease is very rare with an incidence of about 1–2 people per million per year [3]. However, mounting evidence suggests that the proteins implicated in other neurodegenerative diseases, including amyloid beta (Aβ) and tau in Alzheimer’s disease (AD), alpha synuclein (α-syn) in Parkinson’s disease (PD), TAR DNA binding protein (TDP-43) and superoxide dismutase (SOD1) in amyotrophic lateral sclerosis (ALS), and huntingtin (Htt) in Huntington’s disease (HD) spread via prion-like mechanisms [4][5][6][7][8][9][10][11][12].
Another prion-specific concept that has proven applicable to other neurodegenerative diseases is the concept of strains. In prion disease, strains are clinically and pathologically distinct phenotypes thought to arise from different conformations of PrPSc. Each prion strain is associated with a distinct incubation period, rate of disease progression, and symptom profile. Pathologically, strains target different brain regions and have unique PrPSc deposition patterns [13]. Additionally, strains can be characterized biochemically by their variable conformational stability, sensitivity to protease digestion, aggregation kinetics, and glycosylation pattern seen in western blot [14]. Increasingly, researchers have demonstrated that strains account for at least some of the heterogeneity observed in other neurodegenerative diseases, including AD, PD, and ALS [15][16][17][18][19].
The prion-like spreading observed in AD, PD, ALS, frontotemporal dementia (FTD), and HD has allowed for the expansion of prion-specific techniques to study and model these more prevalent diseases. One such technique is the prion organotypic slice culture assay (POSCA), in which organotypic slice cultures are exposed to infectious prions and recapitulate pathology, including strain differences, as seen in vivo [20].
The POSCA was developed in 2008 by Falsig and Aguzzi. They demonstrated that cerebellar organotypic slice cultures from wildtype mice can be infected with multiple prion strains and cultured for up to three months. Proteinase K-resistant PrPSc from the slices is detectable using western blot by 21 days post-infection (dpi) and approaches levels comparable to that seen at terminal in vivo infection by 35 dpi [20]. Real-time quaking-induced conversion (RT-QuIC), a prion amplification technique used to diagnose disease, can detect PrPSc in POSCA as early as seven dpi [21]. Thus, PrPSc accumulation occurs on an accelerated time scale in POSCA compared to in vivo infection. As expected, no PrPSc is observed at any time point in Prnp-knockout slices [20]. The POSCA can also be used as a diagnostic tool for detecting low titres of prion infectivity. For example, cerebellar slice cultures are capable of propagating infection from white blood cells isolated from presymptomatic scrapie-infected sheep [22]. Unlike the scrapie cell assay in which prion infectivity is assessed in neuroblastoma (N2a) or catecholaminergic-derived (CAD) cells, POSCA allows for the study of prion infection in a complex environment in which the in vivo cytoarchitecture is largely preserved. Moreover, organotypic slice cultures from wildtype mice can be infected with many strains, including the mouse-adapted scrapie strain ME7 and the BSE-derived strain 301C, which cannot be propagated in N2a or CAD cells [20]. The widespread availability of transgenic mice further expands the types of prion diseases that can be studied in POSCA. Kondru et al. recently found that cerebellar slices from Tg12 mice, which express elk Prnp, can be infected with and propagate CWD prions [23]. We have also adapted the POSCA cerebellar culture system to coronally sliced whole brain (Figure 1), successfully propagating scrapie strains RML, 22L, and ME7 (Figure 2). This allows examination of patterns of PrP deposition in different brain regions with different strains. Moreover, dividing the sections sagittally allows the use of one hemisphere as an uninfected or untreated control from the same mouse and matched brain region. Alternately, different strains can be applied to matched slices from the same mouse to compare strain propagation. Beyond immunoblotting slice homogenates for PK-resistant PrP, there are a variety of ways to image PrP deposition in the slices, including histoblot [24], heat mapping of total PrP levels (which includes PrPC and PrPSc, Figure 2), or confocal imaging of aggregates of PrP, labelled with antibodies after epitope retrieval [25] (Figure 1). While an immunoblot can demonstrate presence of PrPSc within a slice homogenate, imaging the intact slice allows much more information to be gleaned about the distribution of PrP and pathology.
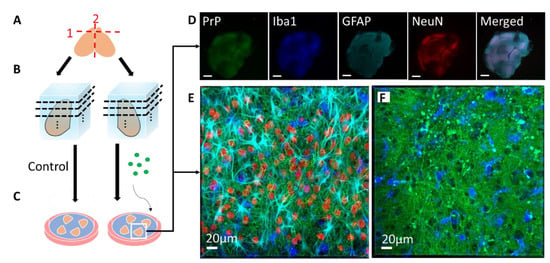
Figure 1. Schematic of coronal slice culture preparation, infection, and confocal imaging. (A) The brain is removed from 8- or 9-day-old mouse pups, the olfactory bulbs are removed, and the brain is hemisectioned sagitally. (B) The hemispheres are embedded in low-melting-point agarose gel and sliced into 275 µm-thick coronal sections with a vibratome. About 40 slices anterior-posterior are obtained from each mouse, with 4 slices cultured per well. Each well is considered a “region”, from 1 (anterior) to 10 (posterior), each representing 1100 µm. (C) The slices are then placed on Milicell cell culture inserts with the culture media below the membrane and can be cultured for as long as three months. During the culture period, genetically identical, location-matched slices can be used as controls or subjected to different treatment conditions in the absence of the blood–brain barrier, such as infection with different prion strains or treatment with different drugs. At various time points, the slices can be immunostained for confocal imaging. (D) Low magnification confocal image of an RML-infected slice that was cultured for 56 days post-infection. PrP (Saf83) is shown in green, microglia (Iba1) are shown in blue, reactive astrocytes (GFAP) are shown in cyan, neuronal nuclei (NeuN) are shown in red. As seen in the NeuN channel, the boundaries of distinct brain regions are discernable (scale = 500 µm). (E) High magnification confocal image of a neocortical region from the RML-infected slice in E, showing the array of cell types and architecture. (F) High magnification confocal image of an RML-infected slice showing PrP aggregates (green) and activated microglia (blue).
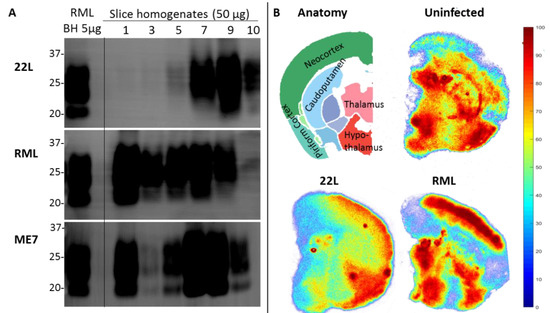
Figure 2. Coronally sliced whole brain prion organotypic slice culture assay (POSCA) can propagate three rodent-adapted scrapie strains. (A) Immunoblots of proteinase K (PK)-digested PrP from slice homogenates of anterior (1) through posterior (10) cultured brain regions. Slices were infected with 22L, RML, or ME7 strain of rodent-adapted scrapie, then harvested at day 56 post-infection. All strains can be propagated in this system. Whether the different levels of PK-resistant PrP are strain-specific is the question of ongoing experiments. POSCA was prepared from C57Bl6 mice for all strains, with the same baseline PrPC levels. An amount of 50 µg total protein before PK digestion was loaded. (B) Anatomy of a slice from region 6 and heat maps for total PrP (PrPC plus PrPSc) of tga20* POSCA uninfected or infected with 22L and RML, showing different distributions of total PrP. Heat map values indicate relative amounts (percentiles of total PrP) within a slice, not absolute values of PrP, so it is the pattern of total PrP distribution, not the intensity of signal, that can be compared between slices. Unlike immunoblots which only provide total levels of a protein from a homogenate, slice culture allows analysis of regional variation within a single slice. * tga20 mice express wildtype mouse PrP at 6× fold higher levels and were the original mouse line used for the first POSCA experiments. PrP antibodies: Sha31 (A), SAF83 (B).
Many pathological features of in vivo prion infection are recapitulated in POSCA. By 42 dpi in RML-infected cerebellar slices, there is significant loss of NeuN and synaptophysin in the granule cell layer compared to non-infected controls, and intense PrP-immunostaining is observed in this region [25][26]. Astrogliosis, increased microglia, and spongiform vacuolation are also observed [24][26], and loss of dendritic spines and changes in spine morphology have been reported by 63 dpi [27]. Interestingly, histoblots show that organotypic slice cultures exhibit strain differences typical of in vivo infection. Diffuse prion deposition is seen in RML strain-infected slices, while the 22L strain induces dense, multifocal plaques, and the 139A strain leads to patchy PrPSc deposition in the white matter [24].
The POSCA has also been used to better understand downstream pathological mechanisms of prion infection. Specifically, Harischandra et al. investigated whether the protein kinase C Cδ (PKCδ), a pro-apoptotic kinase, is involved in prion-induced neuronal death. They infected cerebellar slices from C57BL/6 mice with RML prions. After two weeks, they found more intense PKCδ immunoreactivity in the Purkinje cells of the molecular layer compared to non-infected control slices. They also observed both an increase in PKCδ mRNA and enhanced proteolytic activation of PKCδ in infected slices, suggesting an important role for this kinase in prion-induced cell death [28]. Herrmann et al. found that exposure of cerebellar Tga20 organotypic cultures to RML prions led to significant reactive oxygen species (ROS) production and elevated levels of phosphorylated PERK, eIF2α, and ATF4, which are all markers of endoplasmic reticulum stress. Treatment of slice cultures with ROS scavengers such as ascorbate and isoascorbate was protective, though these compounds did not affect the accumulation of PrPSc [29].
The POSCA is particularly amenable to the study of microglia as they can be suppressed or removed by various methods. Zhu et al. prepared cerebellar slice cultures from tga20/CD11b-thymidine kinase of herpes simplex virus (HSV TK) mice, in which microglia can be conditionally ablated through addition of granciclovir. After infection with RML prions, microglia-depleted slices had significantly fewer granular neurons compared to infected slices in which microglia had been retained [30]. A previous experiment from the same group also showed up to 5x greater PrPSc deposition in microglia-depleted slices [26]. It is also possible to add glial cells to POSCA to study mechanisms of prion spread. Specifically, Hofmann et al. used POSCA to investigate spreading of the N-terminal and middle domain of Sup35 (NM), a yeast prion protein. They infected astrocytes expressing soluble NM with recombinant NM fibrils, which were then added to organotypic mouse hippocampal slices that expressed soluble NM. After 12 days of culture, they found intracellular NM aggregates within the slice [31].
The POSCA has proven useful for testing anti-prion drugs. Cortez et al. found a dose-dependent neuroprotective effect of bile acids tauroursodeoxycholic acid (TUDCA) and ursodeoxycholic acid (UDCA) in RML-infected cerebellar slices from Tga20 mice. Treatment with either compound at seven or 14 dpi led to a significantly lower level of PrPSc without affecting levels of PrPC. Treatment starting at 21 days was not as effective [32]. Margalith et al. tested poly(thiophene-3-acetic acid) (PTAA), a luminescent conjugated polythiophene (LCPs), for its ability to slow prion replication in RML-infected Tga20 cerebellar slice cultures. Interestingly, they observed that PTAA dose-dependently reduced accumulation of PrPSc. However, the resulting PrPSc was actually more resistant to proteolysis [33]. Moreover, Wolf et al. observed that the sulfated polysaccharide DS-500 was effective at reducing PrPSc accumulation and astrocytosis in 22L-infected cerebellar slices compared to control [25]. In another experiment, inhibitors of the mGluR5 receptor, including methyl-6-(phenylethynyl)-pyridine (MPEP), were found to be neuroprotective in RML-infected cerebellar and hippocampal slices. Additionally, knockout of mGluR5 receptors in hippocampal slices reduced cell death in infected slices, confirming the salience of this target for drug intervention [34]. In the CWD-POSCA experiments performed by Kondru et al., seeding activity was significantly reduced by congo red, while quinacrine and astemizole were only mildly effective or ineffective, respectively [23].





