Antisense oligonucleotides (AOs) have been developed to inhibit the production of alternatively spliced carcinogenic isoforms through splice modulation or mRNA degradation. AOs can also be used to induce splice switching, where the expression of an oncogenic protein can be inhibited by the induction of a premature stop codon. In general, AOs are modified chemically to increase their stability and binding affinity. One of the major concerns with AOs is efficient delivery. Strategies for the delivery of AOs are constantly being evolved to facilitate the entry of AOs into cells.
1. Introduction
RNA splicing is a form of RNA processing in which a freshly made precursor messenger RNA (pre-mRNA, or primary transcript) is transformed into a mature mRNA. The process of splicing is catalyzed by the RNA-protein complex known as the spliceosome. The spliceosome consists of five small nuclear ribonucleoproteins (snRNPs), U1, U2, U4, U5 and U6 [1]. During splicing, introns are removed, and exons are joined together.
Splicing can create a range of transcript variants by varying the exon composition of the same transcript. This phenomenon is termed alternative splicing. Alternative splicing is an indispensable process for expanding the spatiotemporal complexity of the transcriptome [2]. More specifically, it is the process wherein the exons of any primary transcript get rearranged to form an mRNA splice variant, and subsequently, the protein isoform, which are distinct both structurally and functionally [3]. In humans, more than 70% of multiexon genes undergo alternative splicing [4]. These splicing events are very tightly regulated and are highly tissue-specific [5]. Different cis- and trans-acting factors regulate the splicing process as they determine the splice site selection [6]. These splice sites can be weakened or strengthened due to point mutations, ultimately changing the splicing events, resulting in disease [6]. Furthermore, in most eukaryotic organisms, there are certain sequences that are ignored by the splicing machinery. These are known as the pseudoexons. In recent years, it has been reported that atypical inclusion of pseudoexons is more frequent than previously understood and is implicated in many human diseases, such as Duchenne muscular dystrophy (DMD) [7].
Cancer is caused by abnormal cell proliferation resulting from altered gene expression [8]. This can include overexpression of oncogenes [9] and production of aberrant splice variants, which subsequently leads to synthesis of carcinogenic protein isoforms [10]. In the past few decades, new developments into understanding the biological basis for cancer progression and therapeutic resistance have led to the identification of many alternative splicing events, which are key in the development of cancer. These carcinogenic splice isoforms are involved in various cellular processes, such as apoptosis, cell signaling and proliferation [11].
Strategies to inhibit the expression of oncogenes and the carcinogenic splice variants of essential genes at the mRNA level have become popular in the past few decades. Pre-mRNA or mRNA with known nucleotide sequences offer a chance to design antisense oligonucleotides (AOs) specific to the transcripts. AOs are short 15–30-base long DNA or RNA molecules that are chemically modified [12]. AOs are designed to be complementary to the target sequence. AOs bind to the target RNA through Watson–Crick base pairing. The specificity of AOs depends on the accuracy of the Watson–Crick base pairing on the estimate that a particular sequence of >16 bases occurs only once in the human genome [13]. Typically, AOs are chemically modified to improve target binding affinity and stability [14][15]. Efficient delivery of AOs into target cells is challenging. Strategies for the delivery of AOs are continually improved to ease the entry of the AOs into the cells [16].
Mechanisms of action of AOs are elucidated in the next section. AOs that induce RNase H-mediated mRNA degradation have been widely used for downregulation of oncogenes [11][17][18]; therefore, this review focuses primarily on six distinct studies that use AO-mediated splice modulation as a therapeutic strategy, with a discussion of different chemical modifications used and delivery strategies employed. In addition, although premature termination of a codon induced by AO-mediated exon skipping has not yet been reported for the development of anticancer AOs, this can potentially be a useful strategy of AO-based cancer therapy (Figure 1).
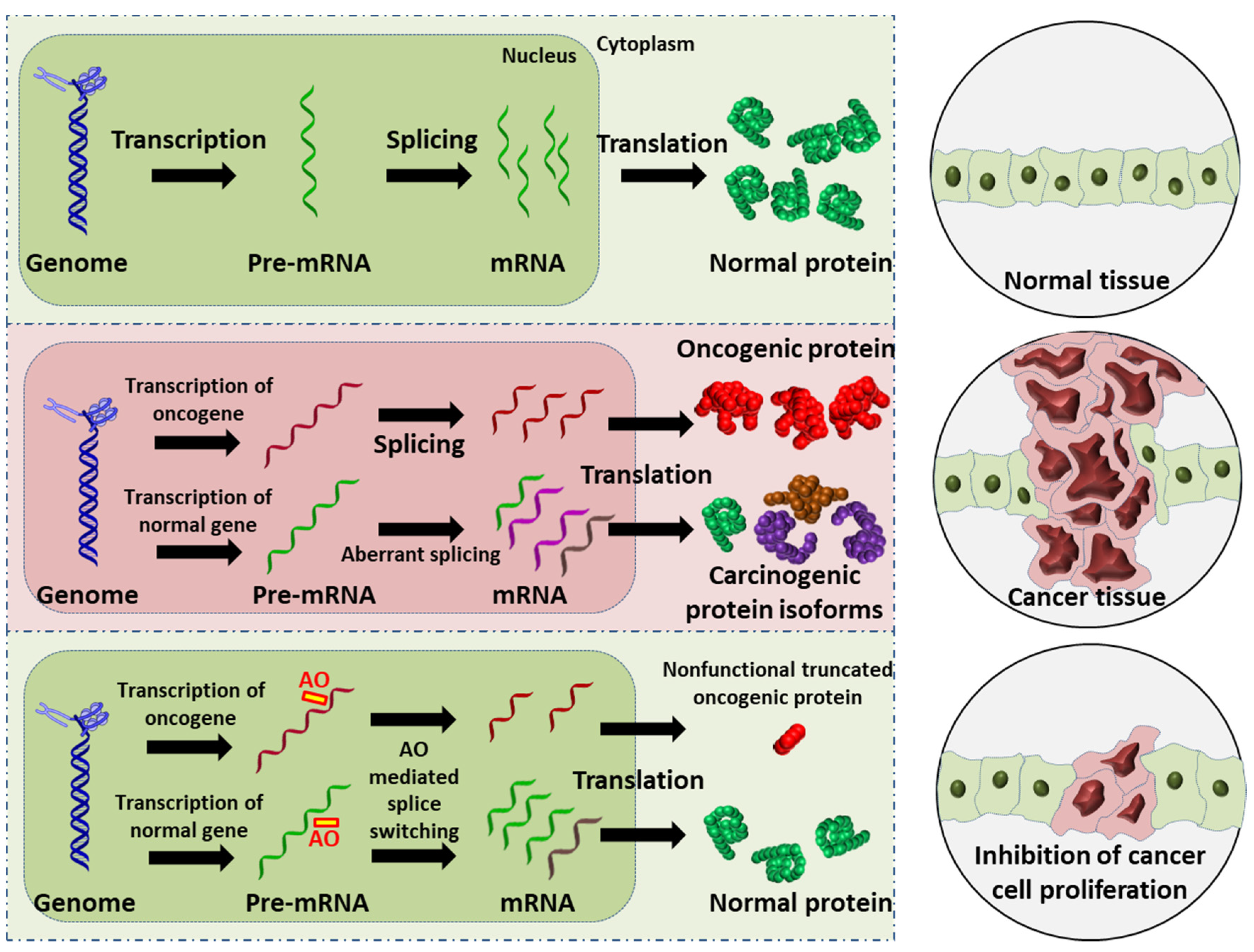 Figure 1. Schematic illustration of the AO-induced anticancer effect. (Upper): normal cell and tissue. (Middle): activation of oncogene leads to production of oncogenic proteins, and the aberrant splicing of normal gene results in splice variants that are translated into carcinogenic protein isoforms; these carcinogenic proteins collectively promote cancer cell proliferation. (Lower): AO-mediated splice switching can not only correct the aberrant splicing, thus reducing the production of carcinogenic protein isoforms (this is the focus of the present review), but can also induce premature stop codons in oncogenic mRNA that results in the synthesis of largely truncated, nonfunctional oncogenic proteins (this is a potential anticancer strategy). Collectively, AO intervention can inhibit cancer cell proliferation.
Figure 1. Schematic illustration of the AO-induced anticancer effect. (Upper): normal cell and tissue. (Middle): activation of oncogene leads to production of oncogenic proteins, and the aberrant splicing of normal gene results in splice variants that are translated into carcinogenic protein isoforms; these carcinogenic proteins collectively promote cancer cell proliferation. (Lower): AO-mediated splice switching can not only correct the aberrant splicing, thus reducing the production of carcinogenic protein isoforms (this is the focus of the present review), but can also induce premature stop codons in oncogenic mRNA that results in the synthesis of largely truncated, nonfunctional oncogenic proteins (this is a potential anticancer strategy). Collectively, AO intervention can inhibit cancer cell proliferation.
2. Exon-Skipping AOs in Cancer
The development of nucleic acid-based therapeutics for cancer has gained prevalence in the past decades. Splice-modulating AOs have been used for suppressing the genes involved in the progression of cancer
[19][20]. In this section the existing exon-skipping antisense oligonucleotide studies are discussed.
2.1. Breast Cancer
In 2018, there were about 2.1 million newly diagnosed breast cancer cases in females worldwide, accounting for about one in four cancer cases in women. Only 5–10% of breast cancers are attributed to hereditary factors
[21]. In general, breast cancers are categorized based on the expression of estrogen receptor, progesterone receptor and the human epidermal growth factor receptor 2 (HER2). Knowing the receptor status of cancer aids in providing specific treatment options. Approximately 25–30% of breast cancers have HER2 overexpression or amplification. Although conventional anti-HER2 therapy has achieved success in treating the HER2+ve subtype of breast cancer, the non-specific action of the HER2 inhibitors has limited the practicality of this approach
[22][23][24]. So far, there are three studies elucidating the use of AO-mediated exon skipping in breast cancer. These studies mainly focus on the genes encoding epidermal growth factor receptors (
EGFR), specifically
HER2 and human epidermal growth factor receptor 4 (
HER4).
A 2′-MOE-modified AO was used to induce skipping of exon 15 in the
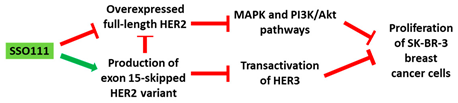
HER2 gene in SK-BR-3 breast cancer cells, leading to downregulation of HER2. Downregulation of HER2 results in upregulation of Δ15HER2, which is an inhibitor of HER2 (
Table 1, No. 1). Δ15HER2 downregulated the full-length HER2 protein and reduced the transphosphorylation of HER3. These AOs served as a platform for the identification of the physiological role of Δ15HER2, a novel HER2 variant. In addition, administration of the AO inhibited the proliferation of the SK-BR-3 cells and induced apoptosis. This suggests that the 2′-MOE AO could be explored in the context of therapeutics
[25]. In another in vitro study, a PNA targeting the intron-exon junction site of exon 19 of HER2 induced exon skipping in SK-BR-3 breast cancer cells and HeLa cervical cancer cell lines (
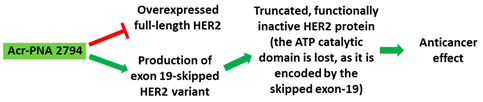
Table 1, No. 2). Exon 19 codes for the ATP catalytic domain of HER2, leading to a functionally inactive HER2 protein in a dominant negative fashion. The PNAs used in this study could be a potential drugs for therapeutic applications
[26]. While current studies have investigated HER2 in a breast cancer context only, the
HER2 oncogene is overexpressed in a variety of additional cancers as well, making a new drug widely applicable
[27].
Table 1. Schematic representation of the six distinct studies on developing splice-modulating AOs targeting cancers. Among these AOs, (1) SSO111, (2) Acr-PNA 2794, (3) SSOe26, and (6) morpholino MDM4 inhibited overexpression of oncogenes by inducing exon skipping, thus producing non-functional variants or leading to nonsense-mediated decay (NMD) of mRNA, while (5) PNA 4577, 4578, 4580 and 4581 predominantly induced intron retention, resulting in the production of non-functional variants. Besides, (4) ASWT1exon5 induced RNase H-mediated degradation of longer transcripts, thus increasing the proportion of naturally occurring, shorter transcripts which exclude an important exon. 2′-MOE, 2′-O-methoxyethyl; 2′-OMe, 2′-O-methyl; PNA, peptide nucleic acid; LNA, locked nucleic acid; PS, phosphorothioate; PMO, phosphorodiamidate morpholino oligomer.
HER4, another member of the EGFR family, plays a diverse role in the development and progression of breast cancer
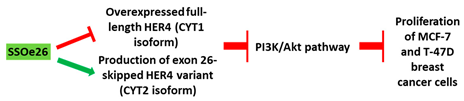
[28]. LNAs targeting the intron-exon junction of exon 26 caused skipping of the exon and switched the splicing of HER4 from cytoplasmic domain 1 (CYT1) isoform to cytoplasmic domain 2 (CYT2) expression in HER4-expressing breast cancer cells (
Table 1, No. 3). These AOs also inhibited the growth of the cancer cells. A similar splice switching pattern was observed in vivo in a xenografted tumor model subcutaneously injected with MCF-7 cells. Furthermore, the AO treatment also reduced tumor growth
[29]. This shows that the use of LNAs targeting exon 26 of HER4 could be explored as a therapeutic strategy.
2.2. Leukemia
As of 2018, the incidence rate of leukemia globally is about 2.4% and the mortality rate about 3.2%
[30]. Treatment options for leukemia vary depending on the type. Apart from conventional treatments like chemotherapy, the use of monoclonal antibodies and gene inhibitors for acute lymphocytic and acute myeloid leukemia are prevalent
[31][32].
Wilms tumor gene (WT1) overexpression is generally a characteristic of acute leukemias and is associated with poor prognosis
[33]. WT1 is located on 11p13 and encodes a zinc finger motif-containing transcription factor. This transcription factor is involved in the regulation of differentiation and growth
[34]. Expression pattern analysis revealed that WT1 is involved in the early stage of hematological cell differentiation
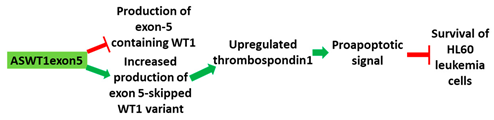
[34]. An AO targeting exon 5 of WT1 showed an upregulation of thrombospondin, a target gene of WT1 in HL-60 cells (
Table 1, No. 4). Upregulation of thrombospondin correlated with cell death in HL-60 cells. K562 cells were also sensitive to the WT1 AOs
[34][35]. WT1 is not only overexpressed in leukemia, but also in a variety of other cancers, including breast cancer, prostate cancer and embryonal cancers
[36]. This suggests exploring the therapeutic benefits of these AOs in leukemia and other cancers.
Terminal deoxynucleotidyl transferase (TdT) is an intracellular protein which is expressed by lymphocytes in the bone marrow and thymus
[37]. High levels of TdT are characteristics of certain lymphoblastic leukemias. TdT is now a well-established diagnostic marker for acute lymphocytic leukaemia
[37][38]. In an in vitro study, 16-mer PNAs were used to target the 5′ and 3′ junctions of intron 7 of TdT in Molt-4 acute lymphoblastic leukemia cells. The specificity of the AO was confirmed by comparing it to a sequence with two base mismatches. Reverse transcriptase PCR (RT-PCR) showed inclusion of intron 7 and subsequently skipping of exon 7 in the TdT gene, leading to significantly reduced TdT expression levels (
Table 1, No. 5). Downregulation of TdT was accompanied with an increase in the rate of apoptosis
[39].
Table 1. Schematic representation of the six distinct studies on developing splice-modulating AOs targeting cancers. Among these AOs, (1) SSO111, (2) Acr-PNA 2794, (3) SSOe26, and (6) morpholino MDM4 inhibited overexpression of oncogenes by inducing exon skipping, thus producing non-functional variants or leading to nonsense-mediated decay (NMD) of mRNA, while (5) PNA 4577, 4578, 4580 and 4581 predominantly induced intron retention, resulting in the production of non-functional variants. Besides, (4) ASWT1exon5 induced RNase H-mediated degradation of longer transcripts, thus increasing the proportion of naturally occurring, shorter transcripts which exclude an important exon. 2′-MOE, 2′-O-methoxyethyl; 2′-OMe, 2′-O-methyl; PNA, peptide nucleic acid; LNA, locked nucleic acid; PS, phosphorothioate; PMO, phosphorodiamidate morpholino oligomer.
| No. |
Research |
Ref. |
| 1 |
AO |
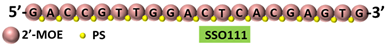
SSO111 is a 20mer fully modified 2′-MOE-PS AO-targeting oncogene HER2. SSO111 induced exon 15 skipping during splicing, leading to the generation of a novel mRNA transcript that excludes exon 15.
 |
[25] |
| Mechanism |
 |
| 2 |
AO |
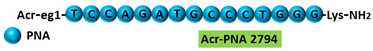
Acr-PNA 2794 is a 15mer fully modified PNA AO conjugated with Acr targeting HER2. Acr-PNA 2794 induced exon-19 skipping, leading to the generation of a novel mRNA transcript that excludes exon-19.
 |
[26] |
| Mechanism |
 |
| 3 |
AO |
SSOe26 is a 15mer LNA-modified mixmer AO targeting HER4. SSOe26 induced exon 26 skipping, leading to the generation of a novel mRNA transcript that excludes exon 26 (CYT2 isoform).
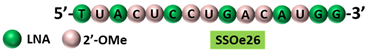 |
[29] |
| Mechanism |
 |
| 4 |
AO |
ASWT1exon5 is a 20mer 2′-MOE-PS gapmer AO targeting oncogene WT1. It induces RNase H-mediated degradation of exon 5-containing transcripts, thus increasing the proportion of transcripts that exclude exon 5.
 |
[35] |
| Mechanism |
 |
| 5 |
AO |
PNA 4577, 4578, 4580, and 4581 are 16mer fully modified PNA AOs conjugated with octaarginine or cholic acid-targeting oncogene TdT. These four PNAs all induced intron 7 retention, leading to the generation of a novel mRNA transcript that included intron-7.
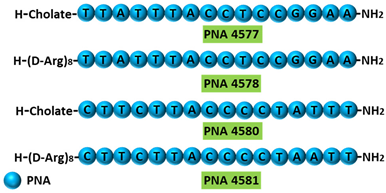 |
[39] |
| Mechanism |
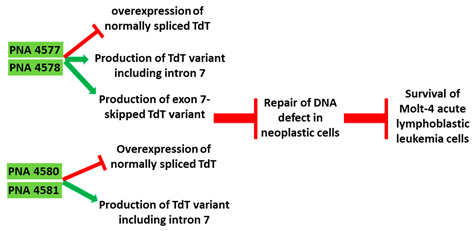 |
| 6 |
AO |
Morpholino MDM4 is a 25mer fully modified PMO AO targeting MDM4. Morpholino MDM4 induced exon 6 skipping, leading to nonsense-mediated decay of the mRNA transcript that excludes exon-6.
 |
[40] |
| Mechanism |
 |
2.3. Melanoma
The global maximum incidence rate and mortality rate of melanoma is about 4.2% and 1.4%, respectively. Although melanomas are not a leading cause of cancer deaths, the incidence rates of melanoma have significantly increased in the past 50 years
[41]. While inactivation of p53 is a common event in most cancers, melanomas with TP53 mutations are very rare. However, the MDM4 regulator of p53 (MDM4) is highly upregulated in most melanomas and promotes the survival of melanoma cells by antagonizing the pro-apoptotic function of p53. This makes MDM4 a key therapeutic target for melanoma
[42]. Overexpression of MDM4 is caused by inclusion of exon 6, which leads to the expression of a full length MDM4. MDM4 exon 6 acts as nonsense-mediated decay (NMD) “NMD switch”
[40]. Though this alternative splicing event is regulated by a number of factors, it was noted that the oncoprotein serine- and arginine-rich splicing factor 3 (SRSF3) is the key initiator of exon 6 inclusion in MDM4
[40]. In this study, a morpholino AO flanking the exon-intron junction of exon 6 and overlapping the SRSF3 binding site inhibited cancer growth both in in vitro and in vivo melanoma models (
Table 1, No. 6). It also sensitized the cells to MAPK-targeting therapeutics, which is a common treatment strategy for melanoma. Similar results were seen in lymphoma models as well. As MDM4 is overexpressed in additional types of cancer, this strategy may be applicable beyond melanoma
[40].
3. Conclusions
The studies reveal that antisense-mediated splice-switching strategy can be effectively used to change the expression of oncogenes and thereby control tumor growth in cancer models. A major drawback in the development of antisense drugs is site-specific delivery and stability. With the advent of new delivery techniques and chemical modifications, this caveat can likely be overcome in the future. Recent studies have also shown that chemically modifying the AOs increased the stability and the binding affinity of the AOs. Furthermore, these AOs were more cost effective with reduced cytotoxicity. This has paved the way for more targets to be explored that underpin a plethora of diseases, taking a step further in finding a possible cure. Antisense technology has progressed rapidly in the past two decades and has now emerged as a highly explored therapeutic strategy that is less or non-toxic and more efficacious.
 +1 point
+1 point





