Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ben Johnson | + 11116 word(s) | 11116 | 2021-09-27 03:30:13 | | | |
| 2 | Bruce Ren | -13 word(s) | 11103 | 2021-10-12 03:44:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Johnson, B. Pre-Clinical Research on MPM Biology. Encyclopedia. Available online: https://encyclopedia.pub/entry/14960 (accessed on 08 February 2026).
Johnson B. Pre-Clinical Research on MPM Biology. Encyclopedia. Available at: https://encyclopedia.pub/entry/14960. Accessed February 08, 2026.
Johnson, Ben. "Pre-Clinical Research on MPM Biology" Encyclopedia, https://encyclopedia.pub/entry/14960 (accessed February 08, 2026).
Johnson, B. (2021, October 11). Pre-Clinical Research on MPM Biology. In Encyclopedia. https://encyclopedia.pub/entry/14960
Johnson, Ben. "Pre-Clinical Research on MPM Biology." Encyclopedia. Web. 11 October, 2021.
Copy Citation
Malignant pleural mesothelioma (MPM) is an aggressive cancer of the lung lining that is associated with asbestos exposure. Due to a lack of effective biomarkers coupled with a long latency period from asbestos exposure to cancer development, prognosis of MPM is poor with an average survival of 8–14 months following diagnosis. Pre-clinical investigations aimed to develop novel biomarkers and treatment strategies are urgently needed to improve MPM diagnosis and treatments available to MPM patients. Novel protein and microRNA biomarkers constitute promising diagnostic biomarkers of MPM; and treatment strategies such as targeted-, immune- and viro-therapy exhibit promising efficacy.
asbestos
malignant pleural mesothelioma
pre-clinical model
biomarker
treatment options
1. Introduction
Malignant pleural mesothelioma (MPM) is a rare, highly aggressive, and incurable cancer of the mesothelium lining the pleural surface of the lungs as a consequence of past exposure to the carcinogen, asbestos. The International Agency for Research on Cancer has established that all fibrous forms of asbestos are carcinogenic to humans and are associated with MPM disease development [1][2]. MPM is the most common type of mesothelioma, accounting for approximately 80% of all mesothelioma cases; with less common forms of mesothelioma affecting the peritoneum, pericardium, and the tunica vaginalis [3]. Although MPM is considered to be rare in comparison to other cancer types, the global incidence of MPM has increased significantly worldwide due to the past widespread and augmented use of asbestos in building materials over the past century. As such, The World Health Organisation estimates that approximately 125 million people are exposed annually worldwide to the carcinogenic mineral fibres in both the workplace and at home; accounting for an estimated 38,000 to 43,000 mesothelioma-related deaths each year [4][5][6]. There are few effective biomarkers and treatment options available to MPM patients in the clinical setting, attributing to a poor associated median survival of only 12–18 months following first-line standard chemotherapy with cisplatin and pemetrexed [7][8]. Patient treatment is further compounded by the fact that MPM is typically associated with a long latency period, with disease symptoms commonly manifesting anywhere between 30–60 years on average following initial asbestos exposure [9][10]. Consequently, a definitive diagnosis of MPM can be challenging and is usually attained upon performing an invasive biopsy procedure and subsequent histological analysis; at which point the disease is often at an advanced stage and treatment with curative intent is largely ineffective [11]. To address this issue, substantial research efforts have been conducted over the past years, having provided valuable insights into the carcinogenic properties of asbestos fibres and their associated molecular alterations; as well as significant pre-clinical studies that have provided the foundation for the potential development of innovative diagnostic and treatment strategies. In particular, pre-clinical research efforts have focused on identifying and evaluating the performance of less-invasive circulating biomarkers, such as blood-based protein or microRNA (miRNA) biomarkers, to assess their potential clinical utility as diagnostic biomarkers of MPM. Alternative pre-clinical studies have focused on the development of innovative MPM-specific treatment strategies, with promising advancements having been made in areas such as targeted therapy, immunotherapy, and virotherapy. Despite these exhaustive research efforts, the precise mechanisms responsible for the genesis of MPM following asbestos exposure are yet to be completely elucidated and consequently, the diagnosis and treatment of MPM ultimately remains ineffective. Hence, continued basic science research involving the use of current and biologically-relevant pre-clinical models is crucially important to mitigate the ongoing asbestos-related disease health burden and the current clinical limitations associated with the diagnosis and treatment of MPM.
This review will summarise the current understanding of the pathogenesis of MPM and associated molecular mechanisms/characteristics, as determined by past pre-clinical research. Furthermore, this article will provide a comprehensive overview of some of the most significant MPM-related pre-clinical studies that have focused on the development of improved diagnostic biomarkers and treatment strategies. As such, research publications relevant to previous and current pre-clinical diagnostic and treatment development strategies for MPM were selected as the inclusion criteria for the purpose of this review. Finally, we conclude by proposing future research directions that could potentially improve the translational potential of prospective MPM-based pre-clinical research.
2. Pre-Clinical Research
Our current knowledge of the aetiology, biological mechanisms, predisposing factors, and potential treatments for MPM has predominantly arisen from basic science or pre-clinical research. Such research has typically involved a mixture of in vitro cell culture-based and in vivo animal-based (predominantly rat and mice) experimental work. The majority of MPM-based studies can best be categorised into three key research themes; namely disease mechanism, diagnosis, and treatment development. This review provides a comprehensive overview of these key research themes and the significant cutting-edge MPM studies that comprise them. Given that MPM is the most commonly/frequently diagnosed form of mesothelioma, the vast majority of past research investigations have predominantly focused on this cancer type. This review will therefore place particular emphasis on research relating to the disease mechanism, diagnosis, and treatment of MPM.
2.1. Disease Mechanism
A number of studies conducted over the past 20 years have led to the identification of dysregulated or aberrant molecular mechanisms that play a role in the development and progression of MPM. These studies have primarily focused on elucidating the chronic inflammatory processes that lead to MPM pathogenesis, as well as the subsequent or predisposing genetic and molecular alterations that lead to disease progression; including alterations to tumour suppressor genes and oncogenes that are unique to MPM tumours. This following section therefore discusses the molecular mechanisms and genetic factors that are known to mediate MPM development and progression.
2.1.1. Pathogenesis
The widely accepted view of MPM pathogenesis is that upon inhalation, the asbestos fibres migrate to the pleura where they directly interact with the human pleural mesothelial cells of the lung and induce a chronic inflammatory response, which over time can lead to malignant transformation [12]. The key contributing molecular processes that have been proposed include: (i) DNA damage induced by reactive oxygen species (ROS) and reactive nitrogen species (RNS) generated by asbestos fibre-exposed mesothelial cells and pleural macrophages attempting to phagocytose the asbestos fibres; (ii) the asbestos fibres absorb a variety of proteins and chemicals, which may result in the accumulation of hazardous carcinogens; and (iii) the asbestos fibre-exposed mesothelial cells and macrophages release cytokines and growth factors, such as high-mobility group box 1 (HMGB1) and tumour-necrosis factor-α (TNF-α), which additionally contribute to chronic inflammation and promote malignant transformation of mesothelial cells that have acquired DNA damage [13][14][15][16][17], as illustrated in Figure 1. This inflammatory-induced malignant transformation can also be attributed to nuclear factor-kappa B (NF-κB) signalling, which augments the survival and proliferation of parietal mesothelial cells [18][19]. Asbestos-induced chronic inflammation also induces aberrant alterations to molecular signalling events, such as aberrant phosphorylation of various protein kinases (e.g., mitogen-activated protein and extracellular signal-regulated kinases 1 and 2), which in turn promotes oncogene activation and loss of tumour suppressor genes; which collectively lead to an induction of abnormal mesothelial cell proliferation and heightened risk of MPM development [20][21]. The most frequently identified molecular alterations to tumour suppressor and oncogenes involved in MPM development are discussed in detail below.
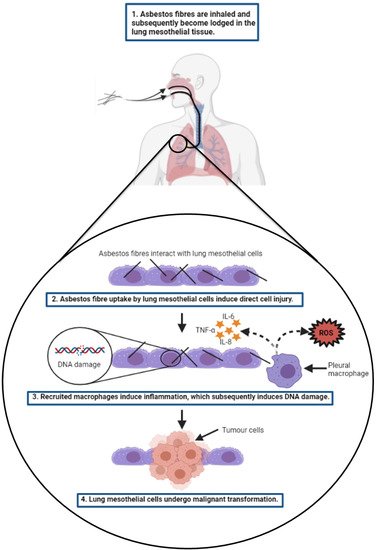
Figure 1. Overview of asbestos-induced mesothelial cell injury leading to the development of malignant pleural mesothelioma (MPM). Asbestos fibres are inhaled and migrate to the pleura of the lungs where they become lodged in the mesothelial tissue. The asbestos fibres induce direct mechanical injury to the mesothelial cells, which induces a chronic inflammation response. This process involves the release of ROS and RNS (not shown) from the iron-containing asbestos fibres and frustrated pleural macrophages that attempt to phagocytose the asbestos fibres. This inflammation process induces cellular DNA damage and aberrant cell signalling, which promotes malignant transformation of the mesothelial cells and cell survival. This ultimately culminates in the development of MPM. All images were created with BioRender.com (accessed on 14 September 2021). Abbreviations: ROS, reactive oxygen species; RNS, reactive nitrogen species; MPM, malignant pleural mesothelioma.
2.1.2. Tumour Suppressor Genes in MPM
Tumour suppressor genes play an important role in cell cycle regulation, normally acting to inhibit cell proliferation, and their inactivation is one of the key molecular events that promote tumour development. The BRCA1-associated protein 1 (BAP1) gene is a tumour suppressor gene located on chromosome 3p21.3 that is commonly found to be inactivated via a germline mutation in individuals with a genetic predisposition for MPM development [13]. BAP1 has been reported to be implicated in crucial biological processes such as cell cycle regulation, DNA damage response, and chromatin dynamics [22]. The multi-domain BAP1 protein, which is encoded by the BAP1 gene, functions as a tumour suppressor deubiquitinating enzyme which regulates gene transcription, cellular differentiation, DNA damage repair, cell metabolism, and apoptosis [23]. In addition to its own tumour suppressor activity, BAP1 induces an accumulation of other DNA-repair proteins at sites of DNA damage [24]. A mutation of the BAP1 gene typically results in a truncated BAP1 protein that is presumed to be degraded prematurely, which impedes its tumour suppressor function [20]. It has been shown that heterozygous germline BAP1 mutations (BAP1 +/−) induce cell metabolism alterations associated with augmented aerobic glycolysis, which in turn induces reprogrammed cellular events that favour carcinogenesis and tumour growth [25]. Aberrant BAP1 expression and somatic truncated BAP1 mutations are typical characteristics of sporadic MPM, with their frequency being variable across different tumour sub-types [26][27]. It has been highlighted that many inactivating mutations occur randomly and are scarcely shared across different MPM biopsies, with the exception of BAP1 which was confirmed to be mutated in 41% and 58% of MPM tumours in two recent next-generation sequencing (NGS) studies conducted by Guo et al. and Lo Iacono et al., respectively; implying that BAP1 is a putative driver mutation for a majority of MPM cases [28][29]. Interestingly, no MPM patients possessing BAP1 germline mutations have been reported to have a known history of occupational exposure to asbestos; indicative that the development of MPM is not directly associated with amount or duration of exposure to asbestos [30]. This has been supported by an in vivo study, whereby it was observed that BAP1 +/− mice were significantly more prone to MPM tumour development following exposure to very low doses of asbestos fibres that have rarely induced MPM in wild-type mice [31]. Moreover, this study demonstrated that the BAP1 +/− mice exposed to low doses of asbestos developed MPM at the same rate as wild-type mice exposed to ten-fold higher doses. These findings have therefore yielded the conclusion that germline BAP1 heterozygosity enhances susceptibility to the carcinogenic effects of low doses of asbestos and has highlighted the potential for the development of therapies designed to restore BAP1 activity.
A high frequency (approximately 25–60%) of somatic BAP1 mutations in MPM has been found to be associated with alterations to other tumour suppressor genes, such as p16/cyclin-dependent kinase inhibitor 2a (CDKN2A), p19/alternate reading frame (ARF) and p15/cyclin-dependent kinase inhibitor 2b (CDKN2B) [32][33]. CDKN2A, CDKN2B, and ARF are present in all healthy (i.e., non-malignant) cells and are essential for normal cell cycle control. CDKN2A in particular, encodes crucial cell cycle regulatory proteins, such as the p16 and p14 ARF proteins, which in turn induces a positive regulation of the p53 tumour suppressor [34]. Additionally, the ARF gene promotes p53-dependent cell apoptosis [35]. Both CDKN2A and CDKN2B encode cyclin-dependent-kinase (CDK) inhibitors, which impede the activity of cyclin-dependent kinase 4 (CDK4) and cyclin-dependent kinase 6 (CDK6). In turn, this contributes to G1-phase cell cycle regulation [36]. CDKN2A, CDKN2B, and ARF have frequently been found to be inactivated by point mutations, aberrant expression, and epigenetic silencing; inducing malignant mesothelial cell transformation upon exposure to asbestos in both in vitro and in vivo models [37][38]. CDKN2A gene deletion occurs in approximately 70–80% of all MPM cases [39][40]. The homozygous deletion of p16/CDKN2A and p19/ARF results in a loss of function of both the p53 and retinoblastoma protein (pRb) tumour suppressors, which consequently leads to a failure of cell cycle arrest [41]. This has been evidenced by a study which showed that the combined inactivation of p16/CDKN2A and p19/ARF expression in vivo was associated with an accelerated initiation of MPM tumourigenesis and reduced survival compared with the inactivation of either gene alone [42].
Neurofibromin 2 (NF2) is an example of another tumour suppressor gene that is frequently inactivated in MPM, as evidenced by either a gene mutation or deletion in up to 38% and 29% of MPM samples, respectively [43][44]. The link between NF2 and MPM tumourigenesis has been exemplified in a study that showed asbestos-treated NF2 +/− mice exhibited an accelerated MPM tumour growth compared to the wild-type controls. Furthermore, biallelic inactivation was detected in all tumours of the NF2 +/− mice, as opposed to 50% of tumours in the wild-type controls [38]. The NF2 gene is located on chromosome 22q12 and encodes the tumour suppressor, merlin, which interacts with a variety of proteins to modulate signal transduction cascades such as mTOR, focal adhesion kinase (FAK), and Hippo signalling pathways. Hence, these pathways have generated considerable research interest for the development of novel targeted therapeutic strategies, which are discussed in a following section of this review.
Less frequent mutations have been detected in MPM biopsies for the tumour protein p53 (TP53) tumour suppressor gene that encodes the p53 transcription factor, with one particular study identifying a somatic TP53 mutation occurrence of up to 17% in MPM patient-derived biopsies [45]. Furthermore, the in vivo deletion of TP53 in mice was found to initiate MPM tumourigenesis, which was associated with secondary BAP1 and NF2 loss [46].
Epigenetic changes in DNA methylation and histone modifications are known to induce the silencing of tumour suppressor genes and promote genomic instability. DNA methylation is a cellular process that involves the covalent addition of methyl groups to the cytosine of CpG dinucleotides that are abundant in CpG islands (CGI). A downregulation of gene expression is associated with DNA methylation, which is mediated by silencing of CGI promotors by DNA-methyltransferase 1 (DNMT1). It has been proposed that asbestos-induced ROS production may promote global hypomethylation in affected mesothelial cells by inducing the expression of the ten-eleven translocation methylcytosine (TET) enzymes, which facilitates the process of active demethylation of hydroxymethylcytosine; thereby evading interference by DNMT1 [47][48]. Global hypomethylation of CpG dinucleotides that do not form CGI has been detected in tumour tissue, whereas hypermethylation has been observed within promotor regions, resulting in aberrant initiation of transcription and genome instability [49][50]. A class of small non-coding RNA’s, microRNA (miRNA), are known epigenetic regulators and they themselves can also be regulated by epigenetic alterations. Both DNA hypomethylation and hypermethylation, as well as histone modifications, play a role in the regulation of miRNA promotor expression. Cancer onset and progression have been linked to epigenetic alterations in tumour suppressor miRNA expression, with a dysregulation and irreversible loss of tumour suppressor miRNA function having been widely reported for MPM [48][51]. The tumour suppressor miRNAs; miR-34, miR-145, and miR-126, have been found to be downregulated in MPM tissues and cell lines due to a mechanism that involves hypermethylation of their promoter regions [52][53][54][55]. Several studies have shown that miR-126 expression is downregulated in tumour tissue in comparison to non-tumour tissue, and its restoration was found to impede tumour cell growth, migration, invasion, and tumourigenesis [56][57][58]. Hypermethylation of the CGI in the epidermal growth factor-like domain-containing protein 7 (EGFL7) intron 2, which contains the transcriptional initiation site of EGFL7 mRNA and miR-126, has been detected in MPM and was found to be significantly associated with poor survival [55]. An inhibition of DNA methylation and histone deacetylation has been shown to be associated with an activation of miR-126 in other non-MPM cancer types, thus providing confirmatory evidence that miR-126 is epigenetically regulated [59][60].
Previous studies have indicated that Poly(ADP-ribose) polymerase-1 (PARP1) is linked to asbestos-induced DNA damage and repair, whereby it has been suggested that PARP1 activity is impeded by asbestos exposure and consequently leads to a high level of DNA instability and induction of malignant transformation [61][62][63]. Despite this, a loss of PARP1 activity does not correlate with a loss in its expression, given that a number of studies have demonstrated an upregulation of PARP1 expression in MPM biospecimens [61][63]. An upregulation of both miR-126 and EGFL7 has been demonstrated in the MPM cell line, H28, upon knocking down PARP1, which was associated with an increase in DNMT1 levels [64]. These results ultimately highlight the involvement of PARP1 and DNMT1 in tumour suppressor miRNA regulation in MPM.
Additional tumour suppressor miRNAs that have been found to be significantly downregulated in MPM biospecimens include those of the miR-15 and miR-16 families. This finding was established upon noting a significant downregulation of miR-15 and miR-16 expression in MPM patient-derived tumour samples and cell lines in comparison to normal mesothelial cells and tissue; whereby a restoration of their expression in both in vitro and in vivo models resulted in an inhibition of MPM cell and tumour growth [65][66]. A similar anti-tumour response has also been reported for the in vitro and in vivo administration of miR-193a-3p mimics; another type of tumour suppressor miRNA which was found to be significantly downregulated in MPM [67]. Collectively, the aforementioned downregulated tumour suppressor miRNAs represent promising candidates for therapeutic manipulation and have therefore garnered increasing research interest in relation to their potential utility as a novel treatment option for MPM, as discussed further in a later section of this review.
The various types of aforementioned tumour suppressor genes that mediate the development and progression of MPM are summarised below in Table 1.
2.1.3. Oncogenes in MPM
The functional role of an oncogene is to promote malignant cell transformation by inducing cell proliferation and inhibiting apoptosis, thus favouring tumour growth and survival. The epidermal growth factor receptor (EGFR) is one such oncogene that is overexpressed in MPM [68]. EGFR is a notoriously known oncogene in many cancer types, and its gene product is a transmembrane glycoprotein belonging to the tyrosine kinase receptor family [20]. The interaction between EGFR and its ligand induces cell proliferation, cell motility and, inhibits apoptosis and the expression of extracellular matrix proteins [69]. The correlation between EGFR over-expression and MPM development has been exemplified by a previous study, which detected an induction of EGFR phosphorylation in asbestos-exposed rat pleural mesothelial cells [70]. The vascular endothelial growth factor (VEGF) and its associated receptor, VEGFR, are also members of the tyrosine kinase family that have been found to be over-expressed in human MPM specimens. Their over-expression in MPM has been shown to play an active role in tumour growth by promoting angiogenesis and lymphangiogenesis [71]. Hence, EGFR and VEGF have attracted considerable research interest for the development of MPM therapeutic strategies that target and inhibit their function [72][73].
A number of upregulated miRNAs that exhibit oncogenic functions, termed oncomiR’s, have been reported for MPM. Two examples include miR-182-5p and miR-183-5p; both having been shown to promote proliferation and invasion in MPM cell lines, with a reduction of these functions being demonstrated upon treatment with miRNA inhibitors [74]. Another example of an miRNA with oncogenic activity in MPM is miR-24-3p, which was found to be overexpressed in both MPM cell lines and tumour samples [75]. This miRNA is known to regulate a number of genes that mediate cell adhesion and communication, and a knockdown of miR-24-3p was shown to impede migration and invasion in both in vitro and in vivo models. Intriguingly, in contrast to the aforementioned upregulated oncogenic miRNA, an oncogenic miRNA, miR-320a, which is downregulated in MPM has been reported recently [76]. The oncogenic status of miR-320a was elucidated after stable ectopic overexpression of this particular miRNA induced an enhanced proliferation and migration ability in the MPM cell line MSTO-211H. Furthermore, it was shown that an induction of p53 over-expression in MPM cell lines was associated with an upregulation of miR-320a and its related miRNAs, miR-200a and miR-34a [76]; both of which are known to target and reduce levels of the programmed death-ligand 1 (PDL-1) [77][78]. An over-expression of PDL-1 in MPM has been linked to its ability to evade an immune-mediated anti-tumour response and is associated with poor patient prognosis [79][80]. Collectively, these findings imply that low expression of miR-320a, miR-34a, and miR-200a in MPM can be attributed to a defective p53-mediated response, which consequently promotes a high expression of PDL-1 and enable MPM cells/tumours to evade immune detection.
The Notch signalling pathway has been identified as an oncogenic mediator of MPM, having been found to be dysregulated in human MPM biopsies [81]. Notch signalling acts as a mediator of short-range cell-to-cell interactions, which involves the regulation of genes controlling cellular processes, such as cell death, proliferation, activation of differentiation, and acquisition of specific cell fates [20]. Increased levels of Notch 1 and reduced levels of Notch 2, encoded by the NOTCH1 and NOTCH2 genes, respectively, have been detected in established biopsy-derived MPM cell lines compared to their normal mesothelial cell counterparts [81]. It has been determined that Notch1 impedes the phosphatase and tensin homolog, PTEN, and activates the PI3K/Akt/mTOR signalling cascade, which promotes growth and survival of MPM tumour cells; indicative of Notch 1′s associated oncogenic role in MPM [81]. Notch 2 however, has been established as a positive transcriptional regulator of PTEN and in contrast to Notch 1, leads to the suppression of PI3K/Akt/mTOR signalling. This has been evidenced by the re-expression of Notch 2 in vitro, which was shown to induce MPM cell cytotoxicity [81]. An inhibition of Notch signalling has been shown to successfully cause a reduction of tumour cell proliferation and inhibition of tumour growth, both in vitro and in vivo, for other non-MPM cancer types [82][83]. Hence, the Notch signalling pathway represents an attractive target for therapeutic intervention of MPM. Despite the known role of Notch signalling in MPM tumourigenesis, combined with the reported efficacy of Notch inhibition in other cancer types, limited research efforts have been conducted in relation to Notch pathway inhibitors and their potential efficacy in MPM.
The different types of aforementioned oncogenes that mediate the development and progression of MPM are summarised below in Table 1.
Table 1. Summary of some of the key genes and miRNAs frequently altered in MPM.
| Gene/miRNA Name | Gene Symbol | Chromosomal Region | General Function | Specific Function | Alteration | Reference |
|---|---|---|---|---|---|---|
| BRCA1-associated protein 1 | BAP1 | 3p21.3 | Tumour suppressor gene | DNA damage repair and cell cycle regulation | Gene deletion | [26][27] |
| p16/cyclin-dependent kinase inhibitor 2A | CDKN2A | 9p21 | Tumour suppressor gene | Cell cycle regulation | Gene deletion | [32][39][40] |
| p15/cyclin-dependent kinase inhibitor 2B | CDKN2B | 9p21 | Tumour suppressor gene | Cell cycle regulation | Gene deletion | [32][33][38] |
| p19/alternate reading frame | ARF | 9p21 | Tumour suppressor gene | Cell cycle regulation | Gene deletion | [32][42] |
| Neurofibromin 2 | NF2 | 22q12 | Tumour suppressor gene | Regulation of cell proliferation, motility, and survival | Gene deletion | [38][43][44] |
| Tumour protein p53 | Tp53 | 17p13.1 | Tumour suppressor gene | Cell cycle regulation | Gene deletion | [45][46] |
| Epidermal Growth Factor Receptor | EGFR | 7p11.2 | Oncogene | Promotes angiogenesis | Overexpression | [68] |
| Vascular Endothelial Growth Factor | VEGF | 6p21.3 | Oncogene | Promotes angiogenesis | Overexpression | [71] |
| Notch homolog 1, translocation-associated (Drosophila) | NOTCH1 | 9q34.3 | Oncogene | Promotes cell proliferation, differentiation and survival | Overexpression | [81] |
| Notch homolog 2 (Drosophila) | NOTCH2 | 1p12 | Tumour suppressor | Suppresses cell proliferation, motility and survival | Underexpression | [81] |
| miR-182-5p, miR-183-5p and miR-24-3p | N/A | N/A | OncomiR’s | Promote cell proliferation, adhesion and invasion | Overexpression | [74][75] |
| miR-320a, miR-34a and miR-200a | N/A | N/A | OncomiR’s | Suppress the expression of PDL-1 | Underexpression | [76] |
2.2. Emerging Biomarkers for the Diagnosis of MPM
The diagnosis of MPM in the clinical setting is challenging. Suspected cases of MPM require sampling of pleural fluid for biochemical and cytological analyses, however, cytological yield is typically low in MPM and biopsies are usually required in order to identify the histological sub-type of the tumour to facilitate a definitive diagnosis [84][85]. Biopsies are obtained through invasive surgeries, such as via a percutaneous needle biopsy procedure or video-assisted thoracoscopic surgery (VATS). The choice of biopsy procedure is highly dependent on the distribution and morphology of the disease, the patient’s suitability for invasive surgery, and the availability of services (e.g., trained staff and resources) [11]. Unfortunately, the VATS procedure is not always feasible to perform on patients who are elderly or of declining health. The use of less-invasive biomarkers has proven to be useful diagnostic tools for other cancers types [86], however, to date, less-invasive biomarkers that have been investigated for MPM are incapable of providing an accurate diagnosis; rendering them unsuitable for clinical implementation. Continued pre-clinical research to identify/validate novel less-invasive biomarkers that are highly sensitive and specific for MPM is greatly warranted and would represent a significant advancement for the diagnosis of MPM. This has therefore become a key priority area of investigation in previous MPM-focused biomarker studies. Some of the most widely studied biomarkers for MPM include mesothelin, megakaryocyte potentiating factor, osteopontin, fibulin-3, high mobility group box 1 (HMGB1), and various miRNA; all of which are summarised below.
Mesothelin is a cell adhesion glycoprotein that is over-expressed in MPM. It has garnered considerable research interest as a potential MPM diagnostic biomarker after it was established that 87% of patients diagnosed with mesothelioma exhibited increased levels of serum mesothelin compared to healthy asbestos-exposed, non-asbestos-exposed individuals and individuals with other malignant or inflammatory lung diseases [87][88]. Follow-up studies have also confirmed the high specificity of effusion-derived mesothelin for MPM, with one particular study reporting a 95% specificity; albeit with a relatively low associated sensitivity of 67% [89]. Less promisingly, results derived from a meta-analysis of data generated from 4491 individuals, consisting of 1026 diagnosed with MPM; reported a sensitivity of only 32% for serum mesothelin, despite a high specificity of 95% [90]. Whilst the diagnostic potential of mesothelin has been determined to be favourable in patients with advanced-stage epithelioid tumour sub-types, it is less useful in sarcomatoid tumour sub-types that rarely express mesothelin [11][90]. Additionally, serum mesothelin levels were found to be increased in only 15% of suspect MPM individuals prior to diagnosis [91]; further indicative of mesothelin’s unsuitability as a biomarker for early detection of MPM. Despite these limitations, the soluble mesothelin-related peptide (SMRP) is still the only blood-based biomarker that has been clinically validated and Food and Drug Administration (FDA)-approved for the monitoring of patients diagnosed with epithelioid or biphasic mesothelioma [92]. Fibulin-3 is another circulating glycoprotein that has garnered considerable research interest as a potential biomarker for MPM, exhibiting a similar specificity to that of mesothelin’s reported value of 95% when measured in plasma, albeit with a lower specificity of just 52% in pleural effusion [93]. Unfortunately, fibulin-3 has been reported to exhibit a poorer associated sensitivity in comparison to soluble mesothelin in terms of providing a reliable MPM diagnosis; yielding a sensitivity of 59% and 22% in pleural effusion- and plasma-derived patient samples, respectively [93]. Rather, fibulin-3 appears to have better potential as a prognostic biomarker after it was established that high levels of fibulin-3 in patient serum correlated with advanced stage MPM [94]. The extracellular cell adhesion protein, osteopontin, has also attracted considerable attention as a potential circulating biomarker for MPM after osteopontin levels were found to be significantly higher in the serum of MPM patients in comparison to healthy asbestos-exposed individuals with a reported specificity and sensitivity of 78% and 86%, respectively [95]. Despite this promising finding, a subsequent study determined that osteopontin is incapable of differentiating between asbestos-induced MPM, pleural metastatic carcinoma, and benign pleural lesion; indicative of its unreliability as an MPM diagnostic biomarker [96]. A recent meta-analysis has further established osteopontin’s unsuitability as a diagnostic biomarker for MPM upon determining that there was no significant difference between the blood osteopontin levels of MPM patients with other cancer type patients [97]. A promising proteomics-based biomarker detection technique, using SOMAscan technology, was utilised to explore the usefulness of a 13-protein biomarker panel, which was able to differentiate between MPM and control samples with a sensitivity and specificity of 93% and 91%, respectively [98]. Furthermore, this 13-protein biomarker was remarkably sensitive for the detection of MPM individuals with varying pathologic stages of disease; with a positive detection of 77%, 93%, 96%, and 96% for stage I, II, III, and IV cases, respectively. These results provide a strong foundation for prospective research focused on assessing their usefulness for surveillance and early diagnosis of MPM in high-risk populations.
As previously discussed, one of the hallmarks of cancer epigenetics is the aberrant global hypomethylation and regional hypermethylation of many tumour suppressor genes. These dysregulated methylated genes have therefore attracted research interest in terms of exploring their utility as potential MPM-specific diagnostic biomarkers. In particular, one promising study has highlighted that three gene loci, Estrogen Receptor 1 (ESR1), Solute Carrier Family 6 Member 20 (SLC6A20), and Spleen Tyrosine Kinase (SYK) have a markedly elevated frequency and/or degree of methylation in MPM in comparison to non-tumour lung tissue. Furthermore, the combination of all three was determined to yield a sensitivity and specificity of 92% and 73%, respectively, upon an evaluation of these gene loci in the aforementioned collection of tissues [99]. These dysregulated genes therefore warrant further investigation to assess their potential as biomarker candidates for MPM detection; particularly prospective studies that aim to assess their specificity and sensitivity using a larger cohort of MPM and non-MPM (other cancer type) specimens, as well as in pleural fluid and/or blood derived from MPM patients.
A class of non-coding RNA, miRNAs, have been reported extensively for many different cancer types, including MPM, however, their clinical potential as diagnostic biomarkers for MPM is still an area undergoing active investigation. Asbestos exposure is known to induce early changes in miRNA expression and therefore a number of miRNA candidates have been proposed as potential biomarkers for early detection of MPM development [100]. Typical miRNA signatures that have been determined for MPM include the overexpression of miR-30b*, miR-197-3p, miR-1281 and miR-32-3p, and deletion of miR-34*, miR-429 and miR-203 [101][102][103]. Additionally, elevated levels of miR-29c* and miR-625-3p have been detected in MPM patient-derived plasma and serum samples compared to those derived from healthy subjects [65], however, it is yet to be determined whether these biomarkers are capable of identifying asbestos-exposed individuals with a prospective MPM diagnosis. Interestingly a number of miRNAs have demonstrated specificity for distinct MPM tumour sub-types. For instance, the expression of miR-135b, miR-181a-2*, miR-499-5p, miR-517b, miR-519d, miR-615-5p, and miR-624 are uniquely characteristic of the epithelioid subtype; the expression of miR-218-2*, miR-346, miR-377*, miR-485-5p, and miR-525-3p are characteristic of the biphasic sub-type; and the expression of miR-301b, miR-433, and miR-543 are characteristic of the sarcomatoid sub-type [101]. Hence, these unique MPM sub-type-specific miRNA signatures present an opportune avenue for research aimed to further validate their usefulness as potential clinical biomarkers for a differential diagnosis of MPM based on sub-type. Additionally, previous research efforts have highlighted the potential of miRNAs to distinguish MPM from other cancer types, such as the miR-200 family in distinguishing MPM from lung adenocarcinomas [104][105]. Using an extensive cohort of lung adenocarcinoma and MPM tissue samples, Benjamin et al. demonstrated that the combination of miR-192, miR-193a-3p, and miR-200c, was able to differentiate between the two malignancies with a specificity and sensitivity of 94% and 100%, respectively [104]. The potential utility of these miRNAs as less-invasive biomarkers of MPM remains to be seen, and prospective studies should address this by assessing their specificity and sensitivity in blood samples obtained from large cohorts of MPM and lung adenocarcinoma patients.
A novel class of non-coding RNA, circular RNAs (circRNAs), constitute a promising blood-based biomarker candidate for early detection of MPM. These differ from linear mRNA, being that they are covalently closed single-stranded RNAs lacking 5′ caps or 3′ poly(A) tails, which are generated from pre-mRNA’s by a process known as backsplicing [106][107]. CircRNAs are highly stable in blood circulation, have a longer associated half-life, and are resistant to exonuclease-mediated degradation compared to their linear counterparts; making them desirable biomarker candidates for blood-based diagnostic applications [108][109][110]. There is growing evidence that indicates their overexpression correlates with tumourigenesis, including malignancies of the lung (including MPM), liver, breast, prostate, bladder, colorectal, ovarian, central nervous system, and stomach; as well as several haematological malignancies [111]. It has been proposed that circRNAs promote tumourigenesis by binding to and impeding the function of tumour-suppressor miRNA; a mechanism coined “miRNA sponging”. Normally miRNAs primarily bind to the 3′ untranslated regions (UTR’s) of specific mRNA targets, whereby they function as post-transcriptional regulators of gene expression for various cellular events, including cell proliferation, migration, differentiation, and apoptosis [112][113]. As a result of recent circRNA profiling, a majority of circRNAs are now known to harbor complementary binding sites for “tumour suppressor” miRNA, implying that they are capable of binding to and inactivating the miRNA; thus impeding their interaction with their mRNA targets [114]. This is supported by growing evidence that have identified a clear trend in over- and under-expression of circRNAs and miRNAs, respectively, in many cancer types, including lung adenocarcinoma, colorectal, gastric, and breast cancer [115]. Whilst circRNA-based biomarker development is still in its infancy, recent related investigations have highlighted the potential for circRNA-based diagnostic and therapeutic strategies to play an important role in cancer management. Such studies include the circRNA profile and bioinformatics analyses carried out by Li et al. and Zhu et al., which have identified two circRNAs as potential biomarkers for early detection of acute myeloid leukaemia and lung adenocarcinoma, respectively [116][117]. CircRNA expression in MPM has been scarcely explored, however, our recent microarray analysis study, conducted on nine different MPM cell lines, revealed upregulated levels for 290 different circRNAs; many of which were found to harbor predicted binding sites for tumour suppressor miRNAs previously shown to be downregulated in MPM tumour samples and cell lines [118]. Hence, these upregulated circRNAs constitute potential biomarker candidates that warrant further investigation using an extensive cohort of MPM patient biospecimens to assess their specificity and sensitivity for MPM detection.
Continued pre-clinical research to identify and validate improved novel biomarkers for their potential application to clinical diagnostics for MPM is urgently needed. However, whilst it is essential to continue pre-clinical research that aims to improve clinical capacity to detect and diagnose suspect patients with MPM, it is equally important to conduct pre-clinical studies that aim to improve and increase the number of therapeutic options available to newly diagnosed MPM patients.
A summary of the aforementioned protein and miRNA biomolecules that have been investigated in relation to their potential as diagnostic biomarkers of MPM is provided below in Table 2.
Table 2. Biomarkers that have been investigated in regards to their diagnostic potential for MPM.
| Biomarker | Biomarker Type | Specificity | Sensitivity | Reference |
|---|---|---|---|---|
| Mesothelin | Protein | High a (95%) | Low b (67%) | [89] |
| Fibulin-3 | Protein | High a (95%) in plasma, but low b (52%) in pleural effusion | Low b (22% and 59% in plasma and pleural effusion, respectively). | [93] |
| Osteopontin | Protein | High a (78%) for differentiating MPM from asbestos-exposed individuals without cancer | High a (86%) for differentiating asbestos-exposed individuals without cancer | |
| SOMAscan 13-protein biomarker panel | Protein | High a (91%) | High a (93%) | [98] |
| ESR1, SLC6A20 and SYK three-gene signature | Methylated genes | Moderate c (73%) | High a (92%) | [99] |
| miR-135b, miR-181a-2 *, miR-499-5p, miR-517b, miR-519d, miR-615-5p and miR-624 | MicroRNA | High a (>75%) for epithelioid subtype MPM. | Not reported | [101] |
| miR-218-2 *, miR-346, miR-377 *, miR-485-5p and miR-525-3p | MicroRNA | High a (>75%) for biphasic subtype MPM | Not reported | [101] |
| miR-301b, miR-433 and miR-543 | MicroRNA | High a (>75%) for sarcomatoid subtype MPM | Not reported | [101] |
| miR-192, miR-193a-3p and miR-200c | MicroRNA | High a (94%) for differentiating MPM from lung adenocarcinoma | High a (100%) for differentiating MPM from lung adenocarcinoma | [104] |
2.3. Treatment Development
2.3.1. Current Standard of Care
The gold standard therapeutic strategies currently available to MPM patients typically involve surgery combined with radiotherapy and/or chemotherapy for resectable tumours, and chemotherapy or radiotherapy only for unresectable tumours. Only a select minority of patients are eligible for a complete tumour resection; either via lung-sacrificing surgery, such as an extrapleural pneumonectomy (EPP), or lung-sparing surgery, such as a pleurectomy/decortication (P/D) [119]. EPP surgery is considerably more beneficial than P/D being that it has an associated average overall survival (OS) benefit of 40 months compared to just 23 months for P/D [120]. Despite the evident OS benefit of EPP surgery, the surgical procedure is high-risk and prone to technical complications such as haemorrhage, empyema, atrial fibrillation, and acute respiratory distress syndrome (ARDS); contributing to a 30-day patient mortality rate of 2–5% after EPP surgery [120]. Patients with unresectable disease are usually treated with palliative systemic chemotherapy, consisting of 4 to 6 cycles of combination chemotherapy with platinum and antifolates [121]. Up until 2020, the FDA- and European Medicines Agency (EMA)-approved cisplatin-pemetrexed combination was the only form of frontline therapy for MPM, having demonstrated a modest increased median survival from 9 to 12 months in most advanced stage MPM patients who are not eligible for surgery [122]. Unfortunately, only around 40% of MPM patients are responsive to this first-line chemotherapy [8]. Recently a new first-line combination therapy regimen, involving the combination of nivolumab and ipilimumab, was approved by the FDA for patients with unresectable MPM, as determined from an open-label, randomised, phase 3 study (CheckMate 743). This new treatment regimen is associated with a median overall patient survival of 18 months, which is a significant improvement to the conventional cisplatin-pemetrexed combination therapy [123]. Despite this recent advancement, there are currently no approved second- or third-line chemotherapy agents available for treatment of MPM and no clear supportive evidence for the use of neoadjuvant or adjuvant chemotherapy [121]. Clearly, there is a desperate need to conduct basic science pre-clinical research that aims to advance and expand therapeutic options for MPM given the limited and modest associated benefits of the current standard of care. To address this unmet need, a number of studies have been carried out in recent years to test and develop innovative therapeutic strategies for MPM. Such novel treatment strategies that have been (and are still being) explored include targeted therapies, immunotherapies, and virotherapies, which are discussed in detail below.
2.3.2. Targeted Therapies
Past research efforts involving a comprehensive genomic profiling of MPM has aided in the detection and characterisation of MPM-specific genetic alterations that represent actionable therapeutic targets for novel MPM treatment strategies. Kinase inhibition, angiogenic inhibition, tumour suppressor genes, and mesothelin are some of the key molecular mechanisms/targets that have been tested for the development of improved MPM treatment strategies.
Receptor tyrosine kinases (RTKs) are a large family of cell cycle regulatory receptors that are commonly over-expressed and activated in MPM [124]. The activation of these receptors induce biochemical signalling cascades that cause the transduction of aberrant cell growth signalling, which ultimately leads to cancer development and progression. Hence, RTKs represent a potential therapeutic target for MPM and therefore this has attracted research aiming to investigate strategies of RTK inhibition and associated anti-tumour activity. MPM tumours secrete pro-angiogenic factors, such as epidermal growth factor (EGF), platelet-derived growth factor (PDGF), and vascular endothelial growth factor (VEGF), all of which are linked to cancer cell proliferation. High levels of EGF, PDGF and VEGF have been reported in serum and pleural effusions derived from MPM patients, and have been linked to poor prognosis and patient survival [125][126][127]. The humanised monoclonal antibody against VEGF, bevacizumab, has demonstrated efficacy and been approved for the treatment of several cancers, such as metastatic renal and colorectal cancer, and non-small cell lung cancer. Unfortunately, clinical trials involving MPM patients treated with bevacizumab have not demonstrated a similar efficacy, whereby the addition of bevacizumab to first-line chemotherapy failed to increase the median survival of MPM patients [128][129][130]; albeit with the exception of a recent phase III trial where bevacizumab-treated patients displayed a modest increase in survival of 2.6 months compared to the standard care treated cohort [7]. Clinical trials involving the use of the imatinib and dasatinib antibody inhibitors against the PDGF beta-receptor (PDGFRβ), which is known to be over-expressed in mesothelioma cell lines, have similarly yielded disappointing results in regards to patient survival [131][132][133][134]. Furthermore, inhibitors of EGFR, such as erlotinib and gefitinib, have also failed in clinical trials despite the overexpression of EGFR in MPM specimens [135][136]. Interestingly, a reduction of angiogenesis and MPM tumour mass and enhanced tumour sensitivity to chemotherapy drug treatment have been demonstrated in the pre-clinical setting, upon the administration of plasminogen activator inhibitor-1 (PAI-1) to mice bearing intrapleural tumours with high VEGF-A expression [137][138]. Therefore, the use of PAI-1 inhibitors represents a promising therapeutic strategy for MPM that warrants further exploration. Another promising therapeutic strategy is the suppression of MPM tumour growth via the inhibition of fibroblast growth factor receptors (FGFRs). Fibroblast growth factor 2 (FGF2) levels have been shown to correlate to tumour aggressiveness and poor patient survival and its down-regulation has been shown to induce the suppression of mesothelioma cell proliferation without affecting the growth of non-malignant cells [139]. An impairment of MPM cell proliferation and migration, both in vitro and in vivo, have been demonstrated upon the inhibition of fibroblast growth factor receptor 1 (FGFR1), as well as enhancing tumour response to chemotherapy drug or ionising irradiation [140]. These studies have therefore demonstrated the potential for the development of novel FGFR-based treatment strategies for MPM and warrant further pre-clinical investigation.
An alternative tyrosine kinase that has been identified as a potential therapeutic target for MPM is the focal adhesion kinase (FAK) protein. FAK, also known as protein tyrosine kinase 2 (PTK2), is located in the cytosol and regulates cellular adhesion, proliferation, migration, and survival; in addition to being critical for cancer stem cell survival and maintenance [121]. In many cancers, FAK overexpression has been linked to aggressive tumour behaviour and promotes immune evasion, resulting in tumour survival and progression [141][142][143][144]. Approximately 35–40% of patients with MPM possess mutations in the neurofibromatosis type 2 (NF2) gene, which encodes the protein Merlin [145]. Functional merlin normally inhibits FAK activation, however it has been demonstrated that in NF2-mutated MPM tumour cells, stable overexpression of FAK induces enhanced tumour invasiveness; which decreased significantly upon restoring Merlin expression [146]. A recognition of the tumour-promoting role of FAK in MPM has prompted the development of FAK small molecule inhibitors and research investigating their potential for use in MPM-based targeted therapies. The small molecule FAK inhibitor, defactinib, yielded promising results in pre-clinical studies in NF2-deficient MPM in vitro and in vivo [147], however, it failed to display any enhanced therapeutic benefit in a subsequent phase II clinical trial in MPM patients and consequently the trial was suspended in late 2015 [121]. An alternative FAK inhibitor, PND-1186 (otherwise known as VS-4718), has demonstrated promising efficacy as a potential treatment option for MPM after it was found that it inhibited proliferation and induced apoptosis in MPM cells lacking Merlin expression [148]. The preferential anti-tumour effect of PND-1186 in Merlin-deficient MPM cells suggests that Merlin is a potential predictive biomarker for an enhanced MPM tumour sensitivity to PND-1186. This postulation is also evidenced by an earlier Phase I clinical trial whereby Merlin-deficient MPM patients treated with the FAK inhibitor, GSK2256098, exhibited an increased progression-free survival (PFS) and disease stabilization [149]. Additionally, the expression of the tumour-suppressor protein, E-cadherin (CDH1), has been identified as another predictive marker of MPM tumour response to FAK inhibition after a study by Kato et al. revealed that a subset of Merlin-deficient MPM cells with E-cadherin-positive expression exhibited resistance to PND-1186 treatment [150]. To address the issue of MPM resistance to FAK inhibitor treatment, prospective pre-clinical studies exploring the efficacy of FAK inhibitor treatments in combination with other functional biomolecules, such as tumour suppressor miRNA mimics, are greatly needed. For instance, a previous bioinformatics study of miRNA expression in MPM has indicated that a number of down-regulated tumour-suppressor miRNAs have a strong link to FAK involvement [151]. The replacement of these down-regulated miRNAs with functional miRNA mimics may potentially be the key to sensitizing E-cadherin-positive MPM to FAK inhibitor treatment.
Tumour suppressor inactivation (i.e., loss-of-function) constitutes one of the most typical mutational events in MPM tumours. BAP1 and CDKN2A are tumour suppressor genes that are frequently inactivated in MPM, and have therefore attracted widespread research interest focused on exploiting these altered genes as potential candidates for the development of targeted therapies [152][153][154][155]. BAP1-mutant MPM cell lines have been shown to be significantly less sensitive than the wild-type counterpart when exposed to the chemotherapy drug, gemcitabine, with significantly less DNA damage being detected in the BAP1-mutant cells [156]. Hence, research efforts have focused on exploring treatment options that can potentially sensitise BAP1-deficient MPM to chemotherapy drug treatment. One particular option that has been investigated is the induction of synthetic lethality of alternate DNA repair pathways, such as through the administration of poly-ADP ribose polymerase (PARP) inhibitors, which have been proven to induce cell death in BAP1-deficient MPM cell lines [61][157]. Promisingly, it has recently been established that the BAP1 mutation status alone does not confer MPM cell sensitivity to PARP inhibition. Rather a combination of both PARP inhibitor and the chemotherapy drug, temozolomide, was found to exhibit efficacy in BAP1-deficient MPM cell lines with an associated high and low expression of Schlafen 11 and O-6-Methylguanine-DNA Methyltransferase (MGMT), respectively [158]. Hence, it has been proposed that such a treatment strategy would be ideal for MPM patients whose tumours possess these combined genetic traits. Furthermore, BAP1 inactivation has been considered to play a key role as an epigenetic regulator associated with the polycomb repressive complex 2 (PRC2) and an enhancer of the zeste homolog 2 (EZH2) pathway. As such, MPM with BAP1 loss have been shown to be sensitised to EZH2 inhibition, both in vitro and in vivo [159]. A phase II trial involving treatment of mesothelioma patients with the EZH2 inhibitor, tazemetostat, demonstrated a promising disease control rate of 51% at 12 weeks [160].
In addition to BAP1-targeted strategies, researchers have investigated therapeutic strategies that are specific to the CDKN2A mutation of MPM tumours. CDKN2A encodes the ADP-ribosylation factor (ARF, commonly known as p14) and INK4A (commonly known as p16), and function by regulating the expression of the p53, and pRB tumour suppressors; ultimately leading to G1 arrest and G0 arrest/apoptosis, respectively [120]. The whole INK4a/ARF locus is typically deleted in more than 70% of human mesothelioma cell lines, which is associated with a loss of function of p53 and pRb, and a consequent failure of cell cycle arrest. It has been shown however, that the restoration of p14ARF via adenoviral p14ARF transfection in human mesothelioma cell lines, induces cell cycle arrest, growth inhibition and apoptosis; which was associated with increased p53 levels and dephosphorylation of pRB [161]. Alternatively, researchers have investigated therapeutic strategies that exploit INK4A and its downstream targets, such as cyclin-dependent kinase 4 (CDK4) and CDK6. The presence of INK4A is associated with an inhibition of CDK4 and CDK6 activity, which leads to cell cycle arrest. In INK4A-deficient MPM cells however, CDK4 and CDK6 remain active and promote cell cycle progression. Therefore researchers have employed small molecule inhibitors of CDK4 and CDK6, such as abemaciclib, to induce apoptosis in MPM tumours and a suppression of tumour growth when used in conjunction with radiation in MPM mouse models [162]. These promising results have led to a current clinical trial (NCT03654833 (MiST)) investigating the efficacy of abemaciclib in p16INK4A-deficient MPM patients. The direct restoration of INK4A via gene therapy has also been investigated in pre-clinical models of mesothelioma and has demonstrated promising anti-tumour activity. In particular, the transduction of MPM cells with the INK4A-expressing adenovirus, Adp16, has been shown to effectively induce cell cycle arrest, reduced cell growth, and eventual cell death [163]. Furthermore, the restoration of INK4A in mesothelioma xenografts was shown to be associated with an inhibition of tumour growth and a reduction in tumour size and spread [163]. Despite these promising results, the re-expression of INK4A via gene therapy has not yet been tested in clinical trials.
Tumour suppressor miRNA have been investigated for their potential role as an MPM targeted therapy option. As discussed previously, a downregulation of miR-16 in MPM cells and tumours is associated with MPM cell/tumour growth. To address this, a restoration of miR-16-5p was investigated both in vitro and in vivo upon reverse transfecting MPM cells with miR-16-5p mimics and administering miR-16-5p-loaded minicells to MPM xenografted tumours, respectively [66]. This study demonstrated that the synthetic restoration of miR-16 in the MPM cells inhibited their growth and sensitised them to chemotherapy drug treatment with pemetrexed and gemcitabine. Furthermore, the intravenous administration of the miR-16-5p-loaded minicells to the tumour-bearing mice resulted in a consistent and dose-dependent inhibition of MPM tumour growth. Additionally, the in vitro and in vivo restoration of the tumour suppressor, miR-193a-3p, displayed similar promising results; inducing an inhibition of MPM cell growth and induction of apoptosis and necrosis in vitro, and inhibition of xenograft tumour growth in vivo [67]. The results from these studies ultimately provided the supportive basis for a phase I clinical trial, MesomiR-1; the first and only in-man miRNA study to date, which investigated the safety and optimal dose of a miR-16-based mimic delivered via anti-EGFR antibody-targeted bacterial minicells, dubbed TargomiR’s. Results generated from this trial validated the safety of the treatment in all 27 patients, with one patient exhibiting an objective response [164] and stable disease in a further 15 patients [165]. Given that only one objective response was attained out of the 27 patients, it has been proposed that a combination treatment approach involving miRNAs combined with other treatment modalities (i.e., chemotherapy) may be necessary in order to elicit an enhanced anti-tumour response [165]. Additionally, the use of alternative miRNA mimics consisting of both two active 5p and 3p arms have been proposed, as well as alternative delivery systems to more accurately deliver the miRNA mimics to MPM tumour cells [166].
Other targeted therapy studies have investigated tumour-specific antigens as potential therapeutic targets for MPM, such as mesothelin. Mesothelin is typically expressed at low levels in most normal tissue types, however, it is overexpressed in a variety of solid tumour types, including MPM [41]. The exact biological function of mesothelin overexpression in MPM is not known, however, pre-clinical evidence suggests that mesothelin plays a role in cell adhesion after it was identified as being the receptor for cancer antigen-125 (CA-125); an interaction which causes heterotypic adhesion and facilitates tumour metastasis [167][168]. For these reasons, mesothelin has been deemed a potential therapeutic target for novel MPM therapies and has attracted considerable research interest. Three different types of mesothelin-targeting agents have been explored, which include anti-tumour antibodies/ antibodies-drug conjugates, mesothelin-targeting vaccines, and mesothelin-targeting recombinant T cells [41]. In particular, the humanised antibody, amatuximab, has shown encouraging anti-tumour activity in combination with chemotherapy drugs against mesothelin-expressing tumours, via inhibition of the interaction between mesothelin and cancer antigen 125 (CA- 125) [169][170]. The immunotoxin, SS1P, consisting of the variable fragment of amatuximab linked to the cytotoxic bacterial toxin Pseudomonas exotoxin A has also exhibited enhanced anti-tumour activity against mesothelin-expressing cancers, including tumour cell lines derived from ascites of patients with peritoneal mesotheliomas [171][172]. Despite this, SS1P has shown only modest activity and responses in treated patients of phase I clinical trials, which has been attributed to the immune-induced formation of neutralizing antibodies acting on SS1P after the first cycle of treatment. To overcome this associated immunogenicity limitation of SS1P, a less immunogenic version (RG7787) has been developed and tested pre-clinically in mesothelin-expressing cancers [173][174]. Alternative mesothelin-specific antibody-drug conjugates that have been investigated in pre-clinical models include αMSLN-MMAE and BAY 94-9343; both of which have exhibited enhanced anti-tumour activity in pre-clinical models compared to standard chemotherapy drug treatment [175][176]. In addition to mesothelin-specific antibodies, genetically modified mesothelin-targeting vaccines have also been investigated for therapeutic potential. In addition to mesothelin-specific antibodies, recent research has focused on vaccine-based targeting of mesothelin-expressing tumour cells. The mesothelin-expressing CRS-207 is a genetically modified Lysteria monocytogenes attenuated vaccine, which has demonstrated therapeutic efficacy in pancreatic tumour-bearing pre-clinical murine models [177]. The in vitro and in vivo pre-clinical testing of CRS-207 in MPM is scarcely reported in the literature, however, a recent clinical-based study has revealed the vaccine has promising therapeutic potential in treated MPM patients; resulting in disease control for 89% of patients and 31% showing a reduction in tumour size [178].
Alternative pre-clinical studies have highlighted argininosuccinate synthetase 1 (ASS1) as a promising targeted therapy candidate for MPM. ASS1 is a rate-limiting enzyme that controls the production of arginine; a known precursor to biomolecules (e.g., nitric oxides and polyamines) that play a role in tumourigenesis [179][180][181]. A study by Szlosarek et al. established that 63% of MPM patient-derived tissue samples exhibited either reduced or absent ASS1 levels. Furthermore, this study demonstrated that ASS1-negative MPM cell lines showed a marked decline in cell viability upon withdrawal of arginine from the culture medium, thus highlighting the potential role of arginine depletion therapy for ASS1-negative MPM patients [182]. This led to a phase II multicentre study which assessed the efficacy of a pegylated arginine deiminase (Adi-PEG 20), an arginine-depleting enzyme, in patients with ASS1-negative MPM. The Adi-PEG 20-treated group exhibited a modest increase in median PFS (3.2 months) in comparison to the group without Adi-PEG 20 treatment (2 months) [183]. Additionally, a phase I dose escalation study demonstrated that the combination of Adi-PEG 20 with cisplatin and pemetrexed was associated with a 78% response rate in a small cohort of ASS1-deficient thoracic cancer patients (including MPM) [184]. A phase 2/3 clinical trial (NCT02709512) is further investigating the efficacy of Adi-PEG 20 treatment in combination with standard cisplatin-pemetrexed therapy in ASS1-negative MPM patients.
The main types of targeted therapies that have been investigated with respect to treatment development for MPM are summarised below in Figure 2. A summary of some of the key clinical trials that have arisen from pre-clinical targeted therapy-based MPM studies are shown in Table 3.
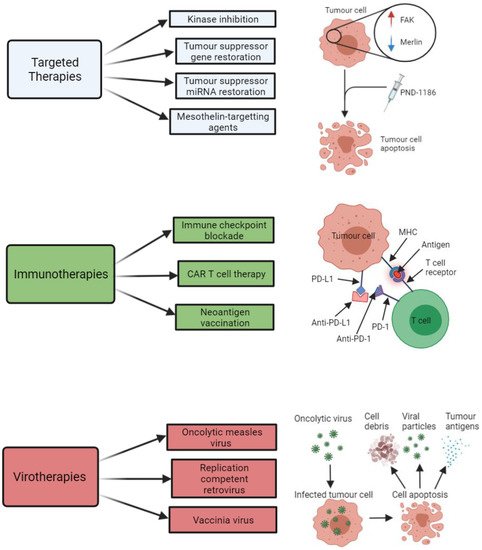
Figure 2. Summary of the key therapeutic strategies that have been explored by researchers investigating potential new treatment options for malignant pleural mesothelioma (MPM). The three key therapeutic strategies include targeted therapies (e.g., kinase inhibitors, such as PND-1186), immunotherapies (e.g., immune checkpoint inhibitors of PD-L1 and PD-1), and virotherapies (e.g., oncolytic measles virus-mediated tumour cell death). All images were created with BioRender.com (accessed on 14 September 2021). Abbreviations: FAK, focal adhesion kinase; miRNA, microRNA; CAR T, chimeric antigen receptor T cell; MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1.
Table 3. Key clinical trials that have arisen from pre-clinical MPM studies.
| Clinical Trial Code | Phase | Title | Treatment | Target | Completed (C) or In Progress (IP) | Outcome | Reference |
|---|---|---|---|---|---|---|---|
| Targeted Therapies | |||||||
| NCT02860286 | II | Study of the EZH2 Inhibitor Tazemetostat in Malignant Mesothelioma | Tazemetostat | BAP1-deficient MPM | C | Disease control was achieved in 51% of patients at 12 weeks and 25% of patients at 24 weeks | [160] |
| NCT00651456 | III | Mesothelioma Avastin Plus Pemetrexed-cisplatin Study (MAPS) | CT + bevacizumab | VEGF | C | 2.6 month increase in patient survival compared to standard CT | [7] |
| NCT03762018 | III | BEAT-meso: Bevacizumab and Atezolizumab in Malignant Pleural Mesothelioma (BEAT-meso) | CT + bevacizumab + atezolizumab | VEGF | IP | - | - |
| NCT03654833 | II | Mesothelioma Stratified Therapy (MiST): A Multi-drug Phase II Trial in Malignant Mesothelioma (MiST) | Abemaciclib | p16INK4A-deficient MPM | IP | - | - |
| NCT02369198 | I | MesomiR 1: A Phase I Study of TargomiRs as 2nd or 3rd Line Treatment for Patients with Recurrent MPM and NSCLC | TargomiRs | EGFR | C | Objective response achieved for 1 patient and stable disease achieved for 15 patients in a cohort of 27 patients | [164][165] |
| NCT01675765 | I | Safety and Efficacy of Listeria in Combination with Chemotherapy as Front-line Treatment for Malignant Pleural Mesothelioma | CT + CRS-207 vaccine | Mesothelin | C | Disease control achieved in 89% of patients, with 31% showing a reduction in tumour size | [178] |
| NCT01279967 | II | A Clinical Trial of ADI-PEG 20TM in Patients with Malignant Pleural Mesothelioma (ADAM) | Adi-PEG 20 | ASS1-deficient MPM | C | 3.2 months PFS achieved for Adi-PEG 20-treated patients compared to 2 months for patients without Adi-PEG 20 treatment | [183] |
| NCT02709512 | II/III | Ph 2/3 Study in Subjects with MPM to Assess ADI-PEG 20 with Pemetrexed and Cisplatin (ATOMIC) | CT + Adi-PEG 20 | ASS1-deficient MPM | IP | - | - |
| Immunotherapies | |||||||
| NCT02054806 | I | Study of Pembrolizumab (MK-3475) in Participants with Advanced Solid Tumors (MK-3475-028/KEYNOTE-28) | Pembrolizumab | PD-1 | C | Objective response was achieved in 28% of patients and stable disease achieved in 48% | [185] |
| NCT02991482 | III | Pembrolizumab Immunotherapy Versus Standard Chemotherapy for Advanced Pre-treated Malignant Pleural Mesothelioma (PROMISE-meso) | CT + pembrolizumab | PD-1 | IP | - | - |
| NCT02899299 | III | Study of Nivolumab Combined with Ipilimumab Versus Pemetrexed and Cisplatin or Carboplatin as First Line Therapy in Unresectable Pleural Mesothelioma Patients (CheckMate743) | CT + nivolumab + ipilimumab | PD-1 and CTLA-4 | IP | A median overall patient survival of 18 months was achieved for patients treated with CT in combination with nivolumab and ipilimumab, compared to 14 months for patients receiving CT alone | [123] |
| NCT01722149 | I | Re-directed T Cells for the Treatment (FAP)-Positive Malignant Pleural Mesothelioma | FAP-specific CAR T cells | FAP | C | - | - |
| Virotherapies | |||||||
| NCT01721018 | I/IIa | Intrapleural Administration of HSV1716 to Treat Patients with Malignant Pleural Mesothelioma. (1716-12) | HSV-1716 | MPM tumour cells | C | A median survival of 15 months and 18 months was achieved in HSV-1716-treated patients and HSV-1716-treated patients that exhibited evidence of anti-tumour immunogenicity. | [186] |
2.3.3. Immunotherapies
There has been growing research interest in immunotherapy as a potential treatment option for MPM after several reported cases of spontaneous regression of MPM identified an associated lymphocyte infiltration in the tumour, which correlated to improved patient survival [187][188][189]. Hence, recent research has focused on strategies to modulate the immune system in order to enhance or facilitate the efficiency of the immune system to elicit an anti-tumour response. Promising types of emerging immunotherapy strategies for MPM include immune checkpoint blockade (ICPB), chimeric antigen receptor (CAR) T cell therapy, T regulatory cell modulation, and neoantigen vaccination.
It has been established that MPM tumours are capable of evading elimination by the immune system; mediated by the involvement of T-cell inhibitory molecules, such as the cytotoxic T-lymphocyte-associated protein 4 (CLTA-4), programmed cell death protein 1 (PD-1), and PD-L1. PD-L1 is known to be expressed in many cancer cells, including MPM, and is associated with a poorer MPM patient median survival of 5 months as opposed to 14.5 months for MPM patients with tumours lacking PD-L1 [79][80]. The binding of PD-L1 to PD-1 on T-cells impedes immune responses against the tumour by inhibiting T-cell proliferation and activation. Thus, tumours that express PD-L1 are capable of evading cytotoxic T-cell activity. Researchers have therefore focused on methods that aim to intervene and disable the interaction between PD-L1 and PD-1, such as through the use of immune checkpoint inhibitors that are able to block this interaction and enable T- and B-cell re-activation [190]. Typical immune checkpoint inhibitors include the monoclonal antibodies tremelimumab, pembrolizumab, and avelumab, which act against CTLA-4, PD-1, and PD-L1, respectively. These antibodies have yielded promising clinical results in melanoma and other cancer types [41][191]. Furthermore, the blockade of the PD-1/PD-L1 interaction and an associated inhibition of tumour growth has been demonstrated in animal models for non-MPM-based studies [192][193]. Limited pre-clinical data has been published for ICPB in MPM however, although a number of ICPB-based clinical studies for MPM have been conducted. The KEYNOTE-28 trial in particular, whereby MPM patients were treated with pembrolizumab, demonstrated some promising results; with up to 28% of patients exhibiting a beneficial response to the drug and another 48% achieving a stable disease response [185]. In contrast, one of the most extensive trials for ICPB in MPM, whereby relapsed MPM patients were treated with tremelimumab, reported that 81% of MPM patients died without a significant difference in overall survival between the drug-treated and placebo-treated cohort [194]. In an effort to improve MPM patient response to ICPB therapy, researchers have resorted to testing the combination of ICPB with other immune-modulatory molecules, targeted therapies, anti-angiogenic agents, chemotherapy drugs, or radiotherapy [195]. The use of immune checkpoint inhibitors in combination with chemotherapy agents has particularly been shown to have enhanced therapeutic benefit in MPM. This was effectively demonstrated in both pre-clinical models and human subjects upon combining the immune checkpoint inhibitor, anti-PD-1, with the chemotherapy drug, gemcitabine, which resulted in an enhanced tumour control and survival outcome when compared to treatment with either agent alone [196]. The combination of anti-PD-1 ICPB with other types of chemotherapy agents, such as cisplatin and paclitaxel, have been shown to exhibit enhanced anti-tumour immunity in other thoracic cancer types, commonly inducing a 50% tumour regression, however, their efficacy is yet to be established for MPM [197]. The use of radiotherapy in combination with ICPB is a promising alternative however, having been investigated in MPM in a recent in vivo study by Wu et al. This study effectively demonstrated a significant inhibition of tumour growth in murine MPM models, which was associated with an increased T cell infiltration into the tumour, upon subjecting them to local radiotherapy in combination with the antibody-mediated blockade of the immune-suppressive cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [198]. The use of tumour suppressor miRNA mimics in combination with ICPB also constitutes a potential therapeutic strategy for MPM that warrants investigation. The rationale for this is based on a previous study by Kao et al., which demonstrated that PD-L1 overexpression in a cohort of 72 patients was associated with a downregulation of tumour suppressor miRNAs miR-15b, miR-16, miR-193a-3p, miR-195, and miR-200c [199]. This same study demonstrated that the treatment of MPM cells with miR-15a and miR-16 mimics led to a downregulation in PD-L1 mRNA and protein levels.
An alternative to the ICPB immunotherapy-based approach that has been explored for MPM is CAR T cell therapy. This form of immunotherapy, based on adoptive cell transfer, involves the generation of T cells that are engineered to recognise specific antigen receptors on the tumour cells. For instance, a study carried out by Klampatsa et al. demonstrated increased cytotoxicity and cytokine release in a panel of four MPM cell lines after patient T-cells were engineered via retroviral transduction to express a panErbB-targeted CAR, co-expressed with a chimeric cytokine receptor in order to promote interleukin-4-mediated CAR T cell proliferation [200]. Alternative studies have employed mesothelin-targeting recombinant T cells to explore their associated therapeutic potential in MPM in both pre-clinical in vitro and in vivo models, having exhibited robust anti-tumour activity in MPM [201][202]. An in vivo study effectively demonstrated the potential efficacy of CAR T cell therapy for MPM, whereby an intrapleural mesothelioma mouse model treated with mesothelin-specific CAR T cells injected into the peritoneum resulted in potent and prolonged anti-tumour immunity [203]. Other targets that have been explored for CAR T cells include components of the stroma of tumours, such as the fibroblast-activating protein (FAP). FAP-specific CAR T cells have exhibited efficacious results in murine subcutaneous MPM models with minimal associated toxicity, which resulted in a phase I clinical trial of human FAP-specific CAR T cells via intrapleural administration in MPM patients [203].
Other researchers are investigating the potential utility of neoantigen vaccination as a means to stimulate the immune system to initiate an immune-mediated anti-tumour response against MPM. The aim of neoantigen vaccination is to prime the host immune system to recognise and target the foreign MPM tumour cells. Neoantigens arise as a result of mutations to oncogenes and tumour suppressor genes, oncogenic viruses, oncofoetal proteins, or overexpression of proteins [120]. They constitute attractive candidates for the development of immune-mediated anti-tumour vaccines given that they; (i) exhibit high binding affinities for T cell and human leukocyte antigen (HLA) receptors, (ii) their expression is restricted only to tumour cells, and (iii) their collective binding affinity and specificity effects enable them to bypass central tolerance and issues associated with autoimmunity [204]. In order to elicit an immune-mediated anti-tumour response, the neoantigens must be presented to T cells upon binding to MHC molecules. Neoantigen vaccination represents a potential form of personalised therapy that is uniquely tailored to individual MPM patients on the basis that their tumour sample can be sequenced using next-generation sequencing (NGS) technology to detect aberrant mutation expression of the neoantigens via RNAseq, and MHC binding potential determined in silico [120][205]. The capacity to identify MPM tumour-specific neoantigens using NGS platforms and capability of vaccinating patients with their own tumour-specific neoantigens has stimulated considerable research interest in anti-cancer vaccination strategies for MPM. One recent study conducted by Sneddon et al. utilised pleural effusion samples collected from 27 mesothelioma patients to screen and identify potential tumour cell-derived neoantigens. A median value of up to 68 different neoantigens was found to be responsive to CD8+ T cells, with a particularly strong CD8+ T cell response being detected for a predicted neoantigen produced by a spontaneous mutation in the ROBO3 gene [206]. This is just one example of a pre-clinical study that provides justified evidence that neoantigens represent promising actionable targets for the development of personalised MPM anti-tumour vaccines. Neoantigen vaccination development for MPM is still in its infancy however, and further pre-clinical research is required to completely elucidate its efficacy and feasibility as a potential new treatment option for MPM patients.
The most common types of immunotherapies that have been investigated in relation to MPM are summarised in Figure 2. A summary of some of the key clinical trials that have arisen from pre-clinical immunotherapy-based MPM studies is shown in Table 3.
2.3.4. Virotherapies
Oncolytic virotherapy is a promising experimental strategy that has been explored as a potential therapeutic option for MPM given that viruses are capable of infecting tumour cells and inducing cell lysis following replication. Oncolytic viruses are capable of inducing host cell death and antigen release; a process which subsequently stimulates the immune system via the activation of both the innate and adaptive immune responses, thus triggering an anti-tumour immune response. MPM tumours are an ideal candidate for assessing the efficacy of viral-induced oncolysis given that the pleural location and (mostly) localised tumour growth allows convenient access to direct intratumoural injection of the virus [207]. Further advantages of using viruses to treat MPM is that they can be engineered to increase their selectivity for tumour cells, as well as being utilised as a means of gene therapy by therapeutically altering the infected tumour cells via gene transfer [207]. A number of different viruses including adenovirus, vesicular stomatis virus, replication-competent retrovirus, measles virus, and the genetic engineered Newcastle disease virus have been assessed in pre-clinical MPM studies with promising results. A recent pre-clinical virotherapy-based study utilised an engineered adenovirus harbouring a deletion in the E1A-Conserved Region 2, to infect and replicate in cancer cells with a defective retinoblastoma pathway, including MPM cells. This adenovirus demonstrated cytotoxicity in vitro and reduced tumour growth in vivo in the MPM cells and a murine xenograft model, respectively [208]. Alternatively, an oncolytic measles virus, genetically modified with an interferon β (IFNβ) gene insertion, was found to induce cell death and retard tumour growth in in vitro and in vivo pre-clinical models of MPM, respectively, which was associated with prolonged survival time [209]. The replication-competent neuroattenuated Herpes Simplex Virus (HSV-1716) has also exhibited potential as a form of oncolytic virotherapy for MPM. The HSV-1716 virus was shown to induce MPM cell cytotoxicity and lysis in vitro, which was associated with reduced tumour growth and significantly prolonged survival in MPM murine models when used in combination with chemotherapy and radiotherapy [210]. This study yielded a promising clinical trial which showed an acceptable safety profile in MPM patients treated intrapleurally with HSV-1716, with more than half of the treated patients displaying evidence of anti-tumour immunogenicity and experiencing prolonged survival, as indicated in Table 3 [211]. Alternatively, a study utilising adenovirus vectors expressing interferon (IFN)-gamma, in combination with the expression of the co-stimulatory molecule CD40L, demonstrated significant suppression of in vivo tumour formation compared to untreated controls [212]. Vaccinia viruses designed to infect tumour cells and elicit an anti-tumour immune response have also exhibited efficacy for treatment of MPM. In particular, the replication-competent GLV-1 h68 virus has been shown to induce lysis in multiple MPM cell lines, which correlated to reduced tumour burden and prolonged survival in MPM murine models following intrapleural delivery of the virus [213].
The key types of virotherapies that have been investigated in relation to MPM are summarised in Figure 2.
References
- Carter, R. IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man Volume 2 Some Inorganic and Organometallic Compounds. J. Clin. Pathol. 1974, 27, 171–172.
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Arsenic, metals, fibres, and dusts. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 11–465.
- Carbone, M.; Ly, B.H.; Dodson, R.F.; Pagano, I.; Morris, P.T.; Dogan, U.A.; Gazdar, A.F.; Pass, H.I.; Yang, H. Malignant mesothelioma: Facts, myths, and hypotheses. J. Cell. Physiol. 2012, 227, 44–58.
- Ezzati, M.; Lopez, A.D.; Rodgers, A.A.; Murray, C.J.L. Comparative Quantification of Health Risks: Global and Regional Burden of Disease Attributable to Selected Major Risk Factors; Ezzati, M., Lopez, A.D., Rodgers, A., Murray, C.J.L., Eds.; World Health Organization: Geneva, Switzerland, 2004.
- Driscoll, T.; Nelson, D.I.; Steenland, K.; Leigh, J.; Concha-Barrientos, M.; Fingerhut, M.; Prüss-Üstün, A. The global burden of disease due to occupational carcinogens. Am. J. Ind. Med. 2005, 48, 419–431.
- Chimed-Ochir, O.; Takahashi, K.; Sorahan, T.; Driscoll, T.; Fitzmaurice, C.; Yoko-O, M.; Sawanyawisuth, K.; Furuya, S.; Tanaka, F.; Horie, S.; et al. 1554 Estimation of the global burden of mesothelioma deaths from incomplete national mortality data. Occup. Environ. Med. 2018, 75, A131.
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A randomised, controlled, open-label, phase 3 trial. Lancet 2015, 387, 1405–1414.
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III Study of Pemetrexed in Combination with Cisplatin Versus Cisplatin Alone in Patients with Malignant Pleural Mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644.
- Bianchi, C.; Giarelli, L.; Grandi, G.; Brollo, A.; Ramani, L.; Zuch, C. Latency periods in asbestos-related mesothelioma of the pleura. Eur. J. Cancer Prev. 1997, 6, 162–166.
- Lanphear, B.P.; Buncher, C.R. Latent period for malignant mesothelioma of occupational origin. J. Occup. Med. 1992, 34, 718–721.
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Talbot, D.C.; Blyth, K.; Maskell, N.A.; Psallidas, I. Malignant pleural mesothelioma: An update on investigation, diagnosis and treatment. Eur. Respir. Rev. 2016, 25, 472–486.
- Kadariya, Y.; Menges, C.W.; Talarchek, J.; Cai, K.Q.; Klein-Szanto, A.J.; Pietrofesa, R.A.; Christofidou-Solomidou, M.; Cheung, M.; Mossman, B.T.; Shukla, A.; et al. Inflammation-Related IL1β/IL1R Signaling Promotes the Development of Asbestos-Induced Malignant Mesothelioma. Cancer Prev. Res. 2016, 9, 406–414.
- Benedetti, S.; Nuvoli, B.; Catalani, S.; Galati, R. Reactive oxygen species a double-edged sword for mesothelioma. Oncotarget 2015, 6, 16848–16865.
- Sekido, Y. Molecular pathogenesis of malignant mesothelioma. Carcinogenesis 2013, 34, 1413–1419.
- Choe, N.; Tanaka, S.; Kagan, E. Asbestos Fibers and Interleukin-1 Upregulate the Formation of Reactive Nitrogen Species in Rat Pleural Mesothelial Cells. Am. J. Respir. Cell Mol. Biol. 1998, 19, 226–236.
- Yang, H.; Rivera, Z.; Jube, S.; Nasu, M.; Bertino, P.; Goparaju, C.; Franzoso, G.; Lotze, M.T.; Krausz, T.; Pass, H.; et al. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 12611–12616.
- Qi, F.; Okimoto, G.; Jube, S.; Napolitano, A.; Pass, H.; Laczko, R.; DeMay, R.M.; Khan, G.; Tiirikainen, M.; Rinaudo, C.; et al. Continuous Exposure to Chrysotile Asbestos Can Cause Transformation of Human Mesothelial Cells via HMGB1 and TNF-α Signaling. Am. J. Pathol. 2013, 183, 1654–1666.
- Yang, H.; Bocchetta, M.; Kroczynska, B.; Elmishad, A.G.; Chen, Y.; Liu, Z.; Bubici, C.; Mossman, B.T.; Pass, H.I.; Testa, J.R.; et al. TNF-α inhibits asbestos-induced cytotoxicity via a NF-κB-dependent pathway, a possible mechanism for asbestos-induced oncogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 10397–10402.
- Sartore-Bianchi, A.; Gasparri, F.; Galvani, A.; Nici, L.; Darnowski, J.W.; Barbone, D.; Fennell, D.A.; Gaudino, G.; Porta, C.; Mutti, L. Bortezomib Inhibits Nuclear Factor-κB–Dependent Survival and Has Potent In vivo Activity in Mesothelioma. Clin. Cancer Res. 2007, 13, 5942–5951.
- Rossini, M.; Rizzo, P.; Bononi, I.; Clementz, A.; Ferrari, R.; Martini, F.; Tognon, M.G. New Perspectives on Diagnosis and Therapy of Malignant Pleural Mesothelioma. Front. Oncol. 2018, 8, 91.
- Robinson, B.W.; Lake, R.A. Advances in malignant mesothelioma. N. Engl. J. Med. 2005, 353, 1591–1603.
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hébert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2013, 111, 285–290.
- Kobrinski, D.A.; Yang, H.; Kittaneh, M. BAP1: Role in carcinogenesis and clinical implications. Transl. Lung Cancer Res. 2020, 9, S60–S66.
- Ismail, I.H.; Davidson, R.; Gagné, J.-P.; Xu, Z.Z.; Poirier, G.G.; Hendzel, M.J. Germline Mutations in BAP1 Impair Its Function in DNA Double-Strand Break Repair. Cancer Res. 2014, 74, 4282–4294.
- Bononi, A.; Yang, H.; Giorgi, C.; Patergnani, S.; Pellegrini, L.; Su, M.; Xie, G.; Signorato, V.; Pastorino, S.; Morris, P.; et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 2017, 24, 1694–1704.
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025.
- Nasu, M.; Emi, M.; Pastorino, S.; Tanji, M.; Powers, A.; Luk, H.; Baumann, F.; Zhang, Y.; Gazdar, A.; Kanodia, S.; et al. High Incidence of Somatic BAP1 alterations in sporadic malignant mesothelioma. J. Thorac. Oncol. 2015, 10, 565–576.
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-Exome Sequencing Reveals Frequent Genetic Alterations in BAP1, NF2, CDKN2A and CUL1 in Malignant Pleural Mesothelioma. Cancer Res. 2015, 75, 264.
- Lo Iacono, M.; Monica, V.; Righi, L.; Grosso, F.; Libener, R.; Vatrano, S.; Bironzo, P.; Novello, S.; Musmeci, L.; Volante, M.; et al. Targeted next-generation sequencing of cancer genes in advanced stage malignant pleural mesothelioma: A retrospective study. J. Thorac. Oncol. 2015, 10, 492–499.
- Carbone, M.; Flores, E.G.; Emi, M.; Johnson, T.A.; Tsunoda, T.; Behner, D.; Hoffman, H.; Hesdorffer, M.; Nasu, M.; Napolitano, A.; et al. Combined Genetic and Genealogic Studies Uncover a Large BAP1 Cancer Syndrome Kindred Tracing Back Nine Generations to a Common Ancestor from the 1700s. PLoS Genet. 2015, 11, e1005633.
- Napolitano, A.; Pellegrini, L.; Dey, A.; Larson, D.; Tanji, M.; Flores, E.G.; Kendrick, B.; Lapid, D.; Powers, A.C.; Kanodia, S.; et al. Minimal asbestos exposure in germline BAP1 heterozygous mice is associated with deregulated inflammatory response and increased risk of mesothelioma. Oncogene 2015, 35, 1996–2002.
- Cheng, J.Q.; Jhanwar, S.C.; Klein, W.M.; Bell, D.W.; Lee, W.C.; Altomare, D.A.; Nobori, T.; Olopade, O.I.; Buckler, A.J.; Testa, J.R. p16 alterations and deletion mapping of 9p21-p22 in malignant mesothelioma. Cancer Res. 1994, 54, 5547–5551.
- Andujar, P.; LeComte, C.; Renier, A.; Fleury-Feith, J.; Kheuang, L.; Daubriac, J.; Janin, A.; Jaurand, M.-C. Clinico-pathological features and somatic gene alterations in refractory ceramic fibre-induced murine mesothelioma reveal mineral fibre-induced mesothelioma identities. Carcinogenesis 2007, 28, 1599–1605.
- Ouelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000.
- Sherr, C.J.; Weber, J.D. The ARF/p53 pathway. Curr. Opin. Genet. Dev. 2000, 10, 94–99.
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55.
- LeComte, C.; Andujar, P.; Renier, A.; Kheuang, L.; Abramowski, V.; Mellotte, L.; Fleury-Feith, J.; Zucman-Rossi, J.; Giovanni, M.; Jaurand, M.-C. Similar Tumor Suppressor Gene Alteration Profiles in Asbestos-Induced Murine and Human Mesothelioma. Cell Cycle 2005, 4, 1862–1869.
- Altomare, D.A.; Vaslet, C.A.; Skele, K.L.; De Rienzo, A.; Devarajan, K.; Jhanwar, S.C.; McClatchey, A.I.; Kane, A.B.; Testa, J.R. A Mouse Model Recapitulating Molecular Features of Human Mesothelioma. Cancer Res. 2005, 65, 8090–8095.
- Husain, A.N.; Colby, T.V.; Ordóñez, N.G.; Allen, T.C.; Attanoos, R.L.; Beasley, M.B.; Butnor, K.J.; Chirieac, L.R.; Churg, A.M.; Dacic, S.; et al. Guidelines for Pathologic Diagnosis of Malignant Mesothelioma 2017 Update of the Consensus Statement From the International Mesothelioma Interest Group. Arch. Pathol. Lab. Med. 2018, 142, 89–108.
- Illei, P.B.; Rusch, V.W.; Zakowski, M.F.; Ladanyi, M. Homozygous Deletion of CDKN2A and Codeletion of the Methylthioadenosine Phosphorylase Gene in the Majority of Pleural Mesotheliomas. Clin. Cancer Res. 2003, 9, 2108–2113.
- Bononi, A.; Napolitano, A.; Pass, H.; Yang, H.; Carbone, M. Latest developments in our understanding of the pathogenesis of mesothelioma and the design of targeted therapies. Expert Rev. Respir. Med. 2015, 9, 633–654.
- Altomare, D.A.; Menges, C.W.; Xu, J.; Pei, J.; Zhang, L.; Tadevosyan, A.; Neumann-Domer, E.; Liu, Z.; Carbone, M.; Chudoba, I.; et al. Losses of Both Products of the Cdkn2a/Arf Locus Contribute to Asbestos-Induced Mesothelioma Development and Cooperate to Accelerate Tumorigenesis. PLoS ONE 2011, 6, e18828.
- Andujar, P.; Pairon, J.-C.; Renier, A.; Descatha, A.; Hysi, I.; Abd-Alsamad, I.; Billon-Galland, M.-A.; Blons, H.; Clin, B.; Danel, C.; et al. Differential mutation profiles and similar intronic TP53 polymorphisms in asbestos-related lung cancer and pleural mesothelioma. Mutagenesis 2013, 28, 323–331.
- Cheng, J.Q.; Lee, W.C.; Klein, M.A.; Cheng, G.Z.; Jhanwar, S.C.; Testa, J.R. Frequent mutations of NF2 and allelic loss from chromosome band 22q12 in malignant mesothelioma: Evidence for a two-hit mechanism of NF2 inactivation. Genes Chromosom. Cancer 1999, 24, 238–242.
- Kim, J.E.; Kim, D.; Hong, Y.S.; Kim, K.-P.; Yoon, Y.K.; Lee, D.H.; Kim, S.-W.; Chun, S.-M.; Jang, S.J.; Kim, T.W. Mutational Profiling of Malignant Mesothelioma Revealed Potential Therapeutic Targets in EGFR and NRAS. Transl. Oncol. 2018, 11, 268–274.
- Krontira, A.; Marazioti, A.; Vreka, M.; Iliopoulou, M.; Lilis, I.; Giotopoulou, G.; Spella, M.; Petrou, H.; Stathopoulos, G. LSC-2017-KRAS & TP53 mutations cause malignant mesothelioma. Eur. Respir. J. 2017, 50, OA293.
- Choudhury, S.R.; Ordaz, J.; Lo, C.L.; Damayanti, N.P.; Zhou, F.; Irudayaraj, J. From the Cover: Zinc oxide Nanoparticles-Induced Reactive Oxygen Species Promotes Multimodal Cyto- and Epigenetic Toxicity. Toxicol. Sci. 2017, 156, 261–274.
- Tomasetti, M.; Gaetani, S.; Monaco, F.; Neuzil, J.; Santarelli, L. Epigenetic Regulation of miRNA Expression in Malignant Mesothelioma: MiRNAs as Biomarkers of Early Diagnosis and Therapy. Front. Oncol. 2019, 9, 1293.
- Chatterjee, R.; Vinson, C. CpG methylation recruits sequence specific transcription factors essential for tissue specific gene expression. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1819, 763–770.
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068.
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016, 1, 15004.
- Tanaka, N.; Toyooka, S.; Soh, J.; Tsukuda, K.; Shien, K.; Furukawa, M.; Muraoka, T.; Maki, Y.; Ueno, T.; Yamamoto, H.; et al. Downregulation of microRNA-34 induces cell proliferation and invasion of human mesothelial cells. Oncol. Rep. 2013, 29, 2169–2174.
- Kubo, T.; Toyooka, S.; Tsukuda, K.; Sakaguchi, M.; Fukazawa, T.; Soh, J.; Asano, H.; Ueno, T.; Muraoka, T.; Yamamoto, H.; et al. Epigenetic Silencing of MicroRNA-34b/c Plays an Important Role in the Pathogenesis of Malignant Pleural Mesothelioma. Clin. Cancer Res. 2011, 17, 4965–4974.
- Cioce, M.; Ganci, F.; Canu, V.; Sacconi, A.; Mori, F.; Canino, C.; Korita, E.; Casini, B.; Alessandrini, G.; Cambria, A.; et al. Protumorigenic effects of mir-145 loss in malignant pleural mesothelioma. Oncogene 2013, 33, 5319–5331.
- Andersen, M.; Trapani, D.; Ravn, J.; Sørensen, J.B.; Andersen, C.B.; Grauslund, M.; Santoni-Rugiu, E. Methylation-associated Silencing of microRNA-126 and its Host Gene EGFL7 in Malignant Pleural Mesothelioma. Anticancer. Res. 2015, 35, 6223–6229.
- Jiang, R.; Zhang, C.; Liu, G.; Gu, R.; Wu, H. Retracted: MicroRNA-126 Inhibits Proliferation, Migration, Invasion, and EMT in Osteosarcoma by Targeting ZEB1. J. Cell. Biochem. 2017, 118, 3765–3774.
- Tomasetti, M.; Nocchi, L.; Staffolani, S.; Manzella, N.; Amati, M.; Goodwin, J.; Kluckova, K.; Nguyen, M.; Strafella, E.; Bajzikova, M.; et al. MicroRNA-126 Suppresses Mesothelioma Malignancy by Targeting IRS1 and Interfering with the Mitochondrial Function. Antioxid. Redox Signal. 2014, 21, 2109–2125.
- Tomasetti, M.; Monaco, F.; Manzella, N.; Rohlena, J.; Rohlenova, K.; Staffolani, S.; Gaetani, S.; Ciarapica, V.; Amati, M.; Bracci, M.; et al. MicroRNA-126 induces autophagy by altering cell metabolism in malignant mesothelioma. Oncotarget 2016, 7, 36338–36352.
- Zhang, Y.; Wang, X.; Xu, B.; Wang, B.; Wang, Z.; Liang, Y.; Zhou, J.; Hu, J.; Jiang, B. Epigenetic silencing of miR-126 contributes to tumor invasion and angiogenesis in colorectal cancer. Oncol. Rep. 2013, 30, 1976–1984.
- Saito, Y.; Friedman, J.M.; Chihara, Y.; Egger, G.; Chuang, J.C.; Liang, G. Epigenetic therapy upregulates the tumor suppressor microRNA-126 and its host gene EGFL7 in human cancer cells. Biochem. Biophys. Res. Commun. 2009, 379, 726–731.
- Pinton, G.; Manente, A.G.; Murer, B.; De Marino, E.; Mutti, L.; Moro, L. PARP1 inhibition affects pleural mesothelioma cell viability and uncouples AKT/mTOR axis via SIRT1. J. Cell. Mol. Med. 2013, 17, 233–241.
- Bhattacharjee, P.; Paul, S. Risk of occupational exposure to asbestos, silicon and arsenic on pulmonary disorders: Understanding the genetic-epigenetic interplay and future prospects. Environ. Res. 2016, 147, 425–434.
- Tomasetti, M.; Amati, M.; Nocchi, L.; Saccucci, F.; Strafella, E.; Staffolani, S.; Tarquini, L.M.; Carbonari, D.; Alleva, R.; Borghi, B.; et al. Asbestos exposure affects poly(ADP-ribose) polymerase-1 activity: Role in asbestos-induced carcinogenesis. Mutagenesis 2011, 26, 585–591.
- Gaetani, S.; Monaco, F.; Alessandrini, F.; Tagliabracci, A.; Sabbatini, A.; Bracci, M.; Valentino, M.; Neuzil, J.; Amati, M.; Santarelli, L.; et al. Mechanism of miR-222 and miR-126 regulation and its role in asbestos-induced malignancy. Int. J. Biochem. Cell Biol. 2020, 121, 105700.
- Kirschner, M.B.; Cheng, Y.Y.; Badrian, B.; Kao, S.C.; Creaney, J.; Edelman, J.J.; Armstrong, N.J.; Vallely, M.P.; Musk, A.W.; Robinson, B.W.S.; et al. Increased circulating miR-625-3p: A potential biomarker for patients with malignant pleural mesothelioma. J. Thorac. Oncol. 2012, 7, 1184–1191.
- Reid, G.; Pel, M.E.; Kirschner, M.B.; Cheng, Y.Y.; Mugridge, N.; Weiss, J.; Williams, M.; Wright, C.; Edelman, J.J.B.; Vallely, M.P.; et al. Restoring expression of miR-16: A novel approach to therapy for malignant pleural mesothelioma. Ann. Oncol. 2013, 24, 3128–3135.
- Williams, M.; Kirschner, M.B.; Cheng, Y.Y.; Hanh, J.; Weiss, J.; Mugridge, N.; Wright, C.; Linton, A.; Kao, S.C.; Edelman, J.J.B.; et al. miR-193a-3p is a potential tumor suppressor in malignant pleural mesothelioma. Oncotarget 2015, 6, 23480–23495.
- Jänne, P.A.; Taffaro, M.L.; Salgia, R.; Johnson, B.E. Inhibition of epidermal growth factor receptor signaling in malignant pleural mesothelioma. Cancer Res. 2002, 62, 5242–5247.
- Ciardiello, F.; Tortora, G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970.
- Faux, S.P.; Houghton, C.E. Cell Signaling in Mesothelial Cells by Asbestos: Evidence for the Involvement of Oxidative Stress in the Regulation of the Epidermal Growth Factor Receptor. Inhal. Toxicol. 2000, 12, 327–336.
- Ohta, Y.; Shridhar, V.; Bright, R.K.; Kalemkerian, G.P.; Du, W.; Carbone, M.; Watanabe, Y.; Pass, H. VEGF and VEGF type C play an important role in angiogenesis and lymphangiogenesis in human malignant mesothelioma tumours. Br. J. Cancer 1999, 81, 54–61.
- Nowak, A.K.; Brosseau, S.; Cook, A.; Zalcman, G. Antiangiogeneic Strategies in Mesothelioma. Front. Oncol. 2020, 10, 126.
- Chia, P.L.; Scott, A.M.; John, T. Epidermal growth factor receptor (EGFR)-targeted therapies in mesothelioma. Expert Opin. Drug Deliv. 2019, 16, 441–451.
- Suzuki, R.; Amatya, V.J.; Kushitani, K.; Kai, Y.; Kambara, T.; Takeshima, Y. miR-182 and miR-183 Promote Cell Proliferation and Invasion by Targeting FOXO1 in Mesothelioma. Front. Oncol. 2018, 8, 446.
- Oliveto, S.; Alfieri, R.; Miluzio, A.; Scagliola, A.; Seclì, R.S.; Gasparini, P.; Grosso, S.; Cascione, L.; Mutti, L.; Biffo, S. A Polysome-Based microRNA Screen Identifies miR-24-3p as a Novel Promigratory miRNA in Mesothelioma. Cancer Res. 2018, 78, 5741–5753.
- Costa, C.; Indovina, P.; Mattioli, E.; Forte, I.M.; Iannuzzi, C.A.; Luzzi, L.; Bellan, C.; De Summa, S.; Bucci, E.; Di Marzo, D.; et al. P53-regulated miR-320a targets PDL1 and is downregulated in malignant mesothelioma. Cell Death Dis. 2020, 11, 748.
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal. 2015, 27, 443–452.
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241.
- Cedrés, S.; Ponce-Aix, S.; Zugazagoitia, J.; Sansano, I.; Enguita, A.; Navarro-Mendivil, A.; Martinez-Marti, A.; Martinez, P.; Felip, E. Analysis of Expression of Programmed Cell Death 1 Ligand 1 (PD-L1) in Malignant Pleural Mesothelioma (MPM). PLoS ONE 2015, 10, e0121071.
- Mansfield, A.S.; Roden, A.C.; Peikert, T.; Sheinin, Y.M.; Harrington, S.M.; Krco, C.J.; Dong, H.; Kwon, E.D. B7-H1 Expression in Malignant Pleural Mesothelioma is Associated with Sarcomatoid Histology and Poor Prognosis. J. Thorac. Oncol. 2014, 9, 1036–1040.
- Graziani, I.; Eliasz, S.; De Marco, M.A.; Chen, Y.; Pass, H.; De May, R.M.; Strack, P.; Miele, L.; Bocchetta, M. Opposite Effects of Notch-1 and Notch-2 on Mesothelioma Cell Survival under Hypoxia Are Exerted through the Akt Pathway. Cancer Res. 2008, 68, 9678–9685.
- Wu, C.X.; Xu, A.; Zhang, C.C.; Olson, P.; Chen, L.; Lee, T.K.; Cheung, T.T.; Lo, C.M.; Wang, X.Q. Notch Inhibitor PF-03084014 Inhibits Hepatocellular Carcinoma Growth and Metastasis via Suppression of Cancer Stemness due to Reduced Activation of Notch1–Stat3. Mol. Cancer Ther. 2017, 16, 1531–1543.
- Mancarella, S.; Serino, G.; Dituri, F.; Cigliano, A.; Ribback, S.; Wang, J.; Chen, X.; Calvisi, D.F.; Giannelli, G. Crenigacestat, a selective NOTCH1 inhibitor, reduces intrahepatic cholangiocarcinoma progression by blocking VEGFA/DLL4/MMP13 axis. Cell Death Differ. 2020, 27, 2330–2343.
- British Thoracic Society Standards of Care Committee. BTS statement on malignant mesothelioma in the UK, 2007. Thorax 2007, 62 (Suppl. S2), ii1–ii19.
- Renshaw, A.A.; Dean, B.R.; Cibas, E.S.; Antman, K.H.; Sugarbaker, D.J. The Role of Cytologic Evaluation of Pleural Fluid in the Diagnosis of Malignant Mesothelioma. Chest 1997, 111, 106–109.
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269.
- Creaney, J.; Yeoman, D.; Demelker, Y.; Segal, A.; Musk, A.; Skates, S.J.; Robinson, B.W. Comparison of Osteopontin, Megakaryocyte Potentiating Factor, and Mesothelin Proteins as Markers in the Serum of Patients with Malignant Mesothelioma. J. Thorac. Oncol. 2008, 3, 851–857.
- Creaney, J.; Robinson, B.W. Detection of Malignant Mesothelioma in Asbestos-Exposed Individuals: The Potential Role of Soluble Mesothelin-Related Protein. Hematol. Clin. N. Am. 2005, 19, 1025–1040.
- Creaney, J.; Segal, A.; Olsen, N.; Dick, I.M.; Musk, A.W.; Skates, S.J.; Robinson, B.W. Pleural Fluid Mesothelin as an Adjunct to the Diagnosis of Pleural Malignant Mesothelioma. Dis. Markers 2014, 2014, 413946.
- Hollevoet, K.; Reitsma, J.B.; Creaney, J.; Grigoriu, B.D.; Robinson, B.W.; Scherpereel, A.; Cristaudo, A.; Pass, H.; Nackaerts, K.; Portal, J.A.R.; et al. Serum Mesothelin for Diagnosing Malignant Pleural Mesothelioma: An Individual Patient Data Meta-Analysis. J. Clin. Oncol. 2012, 30, 1541–1549.
- Creaney, J.; Olsen, N.J.; Brims, F.; Dick, I.M.; Musk, A.W.; de Klerk, N.; Skates, S.J.; Robinson, B.W. Serum Mesothelin for Early Detection of Asbestos-Induced Cancer Malignant Mesothelioma. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2238–2246.
- Ahmadzada, T.; Reid, G.; Kao, S. Biomarkers in malignant pleural mesothelioma: Current status and future directions. J. Thorac. Dis. 2018, 10, S1003–S1007.
- Creaney, J.; Dick, I.M.; Meniawy, T.; Leong, S.L.; Leon, J.S.; Demelker, Y.; Segal, A.; Musk, A.W.; Lee, Y.C.G.; Skates, S.J.; et al. Comparison of fibulin-3 and mesothelin as markers in malignant mesothelioma. Thorax 2014, 69, 895–902.
- Pass, H.I.; Levin, S.M.; Harbut, M.R.; Melamed, J.; Chiriboga, L.; Donington, J.; Huflejt, M.; Carbone, M.; Chia, D.; Goodglick, L.; et al. Fibulin-3 as a Blood and Effusion Biomarker for Pleural Mesothelioma. N. Engl. J. Med. 2012, 367, 1417–1427.
- Pass, H.I.; Lott, D.; Lonardo, F.; Harbut, M.; Liu, Z.; Tang, N.; Carbone, M.; Webb, C.; Wali, A. Asbestos Exposure, Pleural Mesothelioma, and Serum Osteopontin Levels. N. Engl. J. Med. 2005, 353, 1564–1573.
- Grigoriu, B.-D.; Scherpereel, A.; Devos, P.; Chahine, B.; Letourneux, M.; LeBailly, P.; Grégoire, M.; Porte, H.; Copin, M.-C.; Lassalle, P. Utility of Osteopontin and Serum Mesothelin in Malignant Pleural Mesothelioma Diagnosis and Prognosis Assessment. Clin. Cancer Res. 2007, 13, 2928–2935.
- Gillezeau, C.N.; Van Gerwen, M.; Ramos, J.; Liu, B.; Flores, R.; Taioli, E. Biomarkers for malignant pleural mesothelioma: A meta-analysis. Carcinogenesis 2019, 40, 1320–1331.
- Ostroff, R.M.; Mehan, M.R.; Stewart, A.; Ayers, D.; Brody, E.N.; Williams, S.A.; Levin, S.; Black, B.; Harbut, M.; Carbone, M.; et al. Early Detection of Malignant Pleural Mesothelioma in Asbestos-Exposed Individuals with a Noninvasive Proteomics-Based Surveillance Tool. PLoS ONE 2012, 7, e46091.
- Tsou, J.A.; Galler, J.S.; Wali, A.; Ye, W.; Siegmund, K.D.; Groshen, S.; Laird, P.W.; Turla, S.; Koss, M.N.; Pass, H.I.; et al. DNA methylation profile of 28 potential marker loci in malignant mesothelioma. Lung Cancer 2007, 58, 220–230.
- Sage, A.P.; Martinez, V.D.; Minatel, B.C.; Pewarchuk, M.E.; Marshall, E.A.; Macaulay, G.M.; Hubaux, R.; Pearson, D.D.; Goodarzi, A.A.; Dellaire, G.; et al. Genomics and Epigenetics of Malignant Mesothelioma. High-Throughput 2018, 7, 20.
- Guled, M.; Lahti, L.; Lindholm, P.; Salmenkivi, K.; Bagwan, I.; Nicholson, A.G.; Knuutila, S. CDKN2A, NF2, and JUN are dysregulated among other genes by miRNAs in malignant mesothelioma—A miRNA microarray analysis. Genes Chromosom. Cancer 2009, 48, 615–623.
- Taniguchi, T.; Karnan, S.; Fukui, T.; Yokoyama, T.; Tagawa, H.; Yokoi, K.; Ueda, Y.; Mitsudomi, T.; Horio, Y.; Hida, T.; et al. Genomic profiling of malignant pleural mesothelioma with array-based comparative genomic hybridization shows frequent non-random chromosomal alteration regions including JUN amplification on 1p32. Cancer Sci. 2007, 98, 438–446.
- Bononi, I.; Comar, M.; Puozzo, A.; Stendardo, M.; Boschetto, P.; Orecchia, S.; Libener, R.; Guaschino, R.; Pietrobon, S.; Ferracin, M.; et al. Circulating microRNAs found dysregulated in ex-exposed asbestos workers and pleural mesothelioma patients as potential new biomarkers. Oncotarget 2016, 7, 82700–82711.
- Benjamin, H.; Lebanony, D.; Rosenwald, S.; Cohen, L.; Gibori, H.; Barabash, N.; Ashkenazi, K.; Goren, E.; Meiri, E.; Morgenstern, S.; et al. A Diagnostic Assay Based on MicroRNA Expression Accurately Identifies Malignant Pleural Mesothelioma. J. Mol. Diagn. 2010, 12, 771–779.
- Gee, G.; Koestler, D.; Christensen, B.C.; Sugarbaker, D.J.; Ugolini, D.; Ivaldi, G.P.; Resnick, M.B.; Houseman, E.A.; Kelsey, K.T.; Marsit, C.J. Downregulated microRNAs in the differential diagnosis of malignant pleural mesothelioma. Int. J. Cancer 2010, 127, 2859–2869.
- Panda, A.; Grammatikakis, I.; Munk, R.; Gorospe, M.; Abdelmohsen, K. Emerging roles and context of circular RNAs. Wiley Interdiscip. Rev. RNA 2016, 8, e1386.
- Jeck, W.; Sharpless, N. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461.
- Chen, L.-L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 2016, 17, 205–211.
- Zhang, Y.; Xue, W.; Li, X.; Zhang, J.; Chen, S.; Zhang, J.-L.; Yang, L.; Chen, L.-L. The Biogenesis of Nascent Circular RNAs. Cell Rep. 2016, 15, 611–624.
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Pillai, R.S. MicroRNA function: Multiple mechanisms for a tiny RNA? RNA 2005, 11, 1753–1761.
- Haque, S.; Harries, L.W. Circular RNAs (circRNAs) in Health and Disease. Genes 2017, 8, 353.
- Hansen, T.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388.
- Xu, Z.; Yan, Y.; Zeng, S.; Dai, S.; Chen, X.; Wei, J.; Gong, Z. Circular RNAs: Clinical relevance in cancer. Oncotarget 2017, 9, 1444–1460.
- Li, W.; Zhong, C.; Jiao, J.; Li, P.; Cui, B.; Ji, C.; Ma, D. Characterization of hsa_circ_0004277 as a New Biomarker for Acute Myeloid Leukemia via Circular RNA Profile and Bioinformatics Analysis. Int. J. Mol. Sci. 2017, 18, 597.
- Zhu, X.; Wang, X.; Wei, S.; Chen, Y.; Fan, X.; Han, S.; Wu, G. hsa_circ_0013958: A circular RNA and potential novel biomarker for lung adenocarcinoma. FEBS J. 2017, 284, 2170–2182.
- Winata, P.; Cheng, Y.; Williams, M.; Lin, R.C.Y.; Reid, G. Circular RNAs as novel biomarkers for detection of malignant pleural mesothelioma. Asia-Pac. J. Clin. Oncol. 2018, 14, 82–83.
- Nicolini, F.; Bocchini, M.; Bronte, G.; Delmonte, A.; Guidoboni, M.; Crinò, L.; Mazza, M. Malignant Pleural Mesothelioma: State-of-the-Art on Current Therapies and Promises for the Future. Front. Oncol. 2020, 9, 1519.
- Fear, V.S.; Cook, A.M.; Fisher, S.A. The future of mesothelioma research: Basic science research. In Caring for Patients with Mesothelioma: Principles and Guidelines; Springer: Cham, Switzerland, 2019; pp. 203–227.
- Mutti, L.; Peikert, T.; Robinson, B.W.; Scherpereel, A.; Tsao, A.S.; de Perrot, M.; Woodard, G.A.; Jablons, D.M.; Wiens, J.; Hirsch, F.R.; et al. Scientific Advances and New Frontiers in Mesothelioma Therapeutics. J. Thorac. Oncol. 2018, 13, 1269–1283.
- Nowak, A.K. Chemotherapy for malignant pleural mesothelioma: A review of current management and a look to the future. Ann. Cardiothorac. Surg. 2012, 1, 508–515.
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386.
- Brevet, M.; Shimizu, S.; Bott, M.J.; Shukla, N.; Zhou, Q.; Olshen, A.B.; Rusch, V.; Ladanyi, M. Coactivation of Receptor Tyrosine Kinases in Malignant Mesothelioma as a Rationale for Combination Targeted Therapy. J. Thorac. Oncol. 2011, 6, 864–874.
- Strizzi, L.; Catalano, A.; Vianale, G.; Orecchia, S.; Casalini, A.; Tassi, G.; Puntoni, R.; Mutti, L.; Procopio, A. Vascular endothelial growth factor is an autocrine growth factor in human malignant mesothelioma. J. Pathol. 2001, 193, 468–475.
- Filiberti, R.; Marroni, P.; Neri, M.; Ardizzoni, A.; Betta, P.G.; Cafferata, M.A.; Canessa, P.A.; Puntoni, R.; Ivaldi, G.P.; Paganuzzi, M. Serum PDGF-AB in Pleural Mesothelioma. Tumor Biol. 2005, 26, 221–226.
- Betta, P.G.; Libener, R.; Orecchia, S.; Bottero, G.; Paganuzzi, M.; Marroni, P.; Andreatta, R.; Filiberti, R.; Neri, M.; Puntoni, R. Epidermal growth factor in serum from patients with malignant pleural mesothelioma. J. Clin. Oncology. 2004, 22 (Suppl. S14), 7315.
- Ceresoli, G.L.; Zucali, P.A.; Mencoboni, M.; Botta, M.; Grossi, F.; Cortinovis, D.; Zilembo, N.; Ripa, C.; Tiseo, M.; Favaretto, A.G.; et al. Phase II study of pemetrexed and carboplatin plus bevacizumab as first-line therapy in malignant pleural mesothelioma. Br. J. Cancer 2013, 109, 552–558.
- Dowell, J.E.; Dunphy, F.R.; Taub, R.N.; Gerber, D.E.; Ngov, L.; Yan, J.; Xie, Y.; Kindler, H.L. A multicenter phase II study of cisplatin, pemetrexed, and bevacizumab in patients with advanced malignant mesothelioma. Lung Cancer 2012, 77, 567–571.
- Kindler, H.L.; Karrison, T.G.; Gandara, D.R.; Lu, C.; Krug, L.M.; Stevenson, J.P.; Jänne, P.A.; Quinn, D.; Koczywas, M.N.; Brahmer, J.R.; et al. Multicenter, Double-Blind, Placebo-Controlled, Randomized Phase II Trial of Gemcitabine/Cisplatin Plus Bevacizumab or Placebo in Patients with Malignant Mesothelioma. J. Clin. Oncol. 2012, 30, 2509–2515.
- Versnel, M.A.; Claesson-Welsh, L.; Hammacher, A.; Bouts, M.J.; Van Der Kwast, T.H.; Eriksson, A.; Willemsen, R.; Weima, S.M.; Hoogsteden, H.C.; Hagemeijer, A. Human malignant mesothelioma cell lines express PDGF beta-receptors whereas cultured normal mesothelial cells express predominantly PDGF alpha-receptors. Oncogene 1991, 6, 2005–2011.
- Mathy, A.; Baas, P.; Dalesio, O.; van Zandwijk, N. Limited efficacy of imatinib mesylate in malignant mesothelioma: A phase II trial. Lung Cancer 2005, 50, 83–86.
- Tsao, A.S.; Harun, N.; Lee, J.J.; Heymach, J.; Pisters, K.; Hong, W.K.; Fujimoto, J.; Wistuba, I. Phase I Trial of Cisplatin, Pemetrexed, and Imatinib Mesylate in Chemonaive Patients with Unresectable Malignant Pleural Mesothelioma. Clin. Lung Cancer 2013, 15, 197–201.
- Dudek, A.Z.; Pang, H.; Kratzke, R.A.; Otterson, G.A.; Hodgson, L.; Vokes, E.E.; Kindler, H.L. Phase II Study of Dasatinib in Patients with Previously Treated Malignant Mesothelioma (Cancer and Leukemia Group B 30601): A Brief Report. J. Thorac. Oncol. 2012, 7, 755–759.
- Garland, L.L.; Rankin, C.; Gandara, D.R.; Rivkin, S.E.; Scott, K.M.; Nagle, R.B.; Klein-Szanto, A.J.; Testa, J.R.; Altomare, D.A.; Borden, E.C. Phase II Study of Erlotinib in Patients with Malignant Pleural Mesothelioma: A Southwest Oncology Group Study. J. Clin. Oncol. 2007, 25, 2406–2413.
- Govindan, R.; Kratzke, R.A.; Herndon, J.E., 2nd; Niehans, G.A.; Vollmer, R.; Watson, D.; Green, M.R.; Kindler, H.L. Gefitinib in patients with malignant mesothelioma: A phase II study by the Cancer and Leukemia Group B. Clin. Cancer Res. 2005, 11, 2300–2304.
- Takayama, Y.; Hattori, N.; Hamada, H.; Masuda, T.; Omori, K.; Akita, S.; Iwamoto, H.; Fujitaka, K.; Kohno, N. Inhibition of PAI-1 Limits Tumor Angiogenesis Regardless of Angiogenic Stimuli in Malignant Pleural Mesothelioma. Cancer Res. 2016, 76, 3285–3294.
- Hattori, N.; Hamada, H.; Masuda, T.; Akita, S.; Ishikawa, N.; Fujitaka, K.; Haruta, Y.; Murai, H.; Kohno, N. SK-216, An Inhibitor Of Plasminogen Activator Inhibitor-1, Inhibits Tumor Growth In A Mouse Model Of Pleural Mesothelioma. D23 Moonraker: Diagnosis and Management of the Pleural Space. Am. J. Respir. Crit. Care Med. 2014, 189, A5487.
- Stapelberg, M.; Gellert, N.; Swettenham, E.; Tomasetti, M.; Witting, P.K.; Procopio, A.; Neuzil, J. Alpha-tocopheryl succinate inhibits malignant mesothelioma by disrupting the fibroblast growth factor autocrine loop: Mechanism and the role of oxidative stress. J. Biol. Chem. 2005, 280, 25369–25376.
- Schelch, K.; Hoda, M.A.; Klikovits, T.; Münzker, J.; Ghanim, B.; Wagner, C.; Garay, T.; Laszlo, V.; Setinek, U.; Dome, B.; et al. Fibroblast Growth Factor Receptor Inhibition Is Active against Mesothelioma and Synergizes with Radio- and Chemotherapy. Am. J. Respir. Crit. Care Med. 2014, 190, 763–772.
- Shanthi, E.; Krishna, M.H.; Arunesh, G.M.; Reddy, K.V.; Kumar, J.S.; Viswanadhan, V.N. Focal adhesion kinase inhibitors in the treatment of metastatic cancer: A patent review. Expert Opin. Ther. Patents 2014, 24, 1077–1100.
- Golubovskaya, V.M.; Cance, W.G. Focal Adhesion Kinase and p53 Signaling in Cancer Cells. Int. Rev. Cytol. 2007, 263, 103–153.
- Moen, I.; Gebre, M.; Alonso-Camino, V.; Chen, D.; Epstein, D.; McDonald, D.M. Anti-metastatic action of FAK inhibitor OXA-11 in combination with VEGFR-2 signaling blockade in pancreatic neuroendocrine tumors. Clin. Exp. Metastasis 2015, 32, 799–817.
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860.
- Schunselaar, L.M.; Quispel-Janssen, J.M.; Neefjes, J.; Baas, P. A catalogue of treatment and technologies for malignant pleural mesothelioma. Expert Rev. Anticancer Ther. 2016, 16, 455–463.
- Poulikakos, P.I.; Xiao, G.-H.; Gallagher, R.; Jablonski, S.; Jhanwar, S.C.; Testa, J.R. Re-expression of the tumor suppressor NF2/merlin inhibits invasiveness in mesothelioma cells and negatively regulates FAK. Oncogene 2006, 25, 5960–5968.
- Pachter, J.A.; Kolev, V.N.; Schunselaar, L.; Shapiro, I.M.; Bueno, R.; Baas, P.; Xu, Q.; Weaver, D.T. Abstract 4236: FAK inhibitor VS-6063 (defactinib) targets mesothelioma cancer stem cells, which are enriched by standard of care chemotherapy. Cancer Res. 2015, 75, 4236.
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin Deficiency Predicts FAK Inhibitor Sensitivity: A Synthetic Lethal Relationship. Sci. Transl. Med. 2014, 6, 237ra68.
- Soria, J.C.; Gan, H.K.; Blagden, S.P.; Plummer, R.; Arkenau, H.T.; Ranson, M.; Evans, T.R.J.; Zalcman, G.; Bahleda, R.; Hollebecque, A.; et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 2268–2274.
- Kato, T.; Sato, T.; Yokoi, K.; Sekido, Y. E-cadherin expression is correlated with focal adhesion kinase inhibitor resistance in Merlin-negative malignant mesothelioma cells. Oncogene 2017, 36, 5522–5531.
- Sarun, K.C.Y.; Schelch, K.; Reid, R. Combination of MicroRNAs provide a synergistic effect on growth inhibition on MPM cells. In Proceedings of the COSA’s 45th Annual Scientific Meeting, Mesothelioma and Gastro-Intestinal Cancers: Technology and Genomics, Perth, Australia, 13–15 November 2018.
- Yoshikawa, Y.; Emi, M.; Hashimoto-Tamaoki, T.; Ohmuraya, M.; Sato, A.; Tsujimura, T.; Hasegawa, S.; Nakano, T.; Nasu, M.; Pastorino, S.; et al. High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc. Natl. Acad. Sci. USA 2016, 113, 13432–13437.
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672.
- Betti, M.; Aspesi, A.; Biasi, A.; Casalone, E.; Ferrante, D.; Ogliara, P.; Gironi, L.C.; Giorgione, R.; Farinelli, P.; Grosso, F.; et al. CDKN2A and BAP1 germline mutations predispose to melanoma and mesothelioma. Cancer Lett. 2016, 378, 120–130.
- Serrano, M.; Lee, H.-W.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R. Role of the INK4a Locus in Tumor Suppression and Cell Mortality. Cell 1996, 85, 27–37.
- Guazzelli, A.; Meysami, P.; Bakker, E.; Demonacos, C.; Giordano, A.; Krstic-Demonacos, M.; Mutti, L. BAP1 Status Determines the Sensitivity of Malignant Mesothelioma Cells to Gemcitabine Treatment. Int. J. Mol. Sci. 2019, 20, 429.
- Srinivasan, G.; Sidhu, G.S.; Williamson, E.A.; Jaiswal, A.S.; Najmunnisa, N.; Wilcoxen, K.; Jones, D.; George, T.J.; Hromas, R. Synthetic lethality in malignant pleural mesothelioma with PARP1 inhibition. Cancer Chemother. Pharmacol. 2017, 80, 861–867.
- Rathkey, D.; Khanal, M.; Murai, J.; Zhang, J.; Sengupta, M.; Jiang, Q.; Morrow, B.; Evans, C.N.; Chari, R.; Fetsch, P.; et al. Sensitivity of Mesothelioma Cells to PARP Inhibitors Is Not Dependent on BAP1 but Is Enhanced by Temozolomide in Cells with High-Schlafen 11 and Low-O6-methylguanine-DNA Methyltransferase Expression. J. Thorac. Oncol. 2020, 15, 843–859.
- LaFave, L.M.; Béguelin, W.; Koche, R.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Konstantinoff, K.; et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 2015, 21, 1344–1349.
- Zauderer, M.G.; Szlosarek, P.; Le Moulec, S.; Popat, S.; Taylor, P.; Planchard, D.; Scherpereel, A.; Jahan, T.; Koczywas, M.; Forster, M.; et al. Phase 2, multicenter study of the EZH2 inhibitor tazemetostat as monotherapy in adults with relapsed or refractory (R/R) malignant mesothelioma (MM) with BAP1 inactivation. J. Clin. Oncol. 2018, 36, 8515.
- Yang, C.-T.; You, L.; Yeh, C.-C.; Chang, J.W.-C.; Zhang, F.; McCormick, F.; Jablons, D.M. Adenovirus-mediated p14(ARF) gene transfer in human mesothelioma cells. J. Natl. Cancer Inst. 2000, 92, 636–641.
- Matsumoto, S.; Fukuda, A.; Nakamichi, T.; Nakamura, A.; Kuroda, A.; Hashimoto, M.; Takuwa, T.; Kondo, N.; Hasegawa, S. CDK4/6 inhibitor and radiation therapy in malignant pleural mesothelioma. J. Clin. Oncol. 2018, 36 (Suppl. S15), e24326.
- Frizelle, S.P.; Grim, J.; Zhou, J.; Gupta, P.; Curiel, D.T.; Geradts, J.; Kratzke, R.A. Re-expression of p16INK4a in mesothelioma cells results in cell cycle arrest, cell death, tumor suppression and tumor regression. Oncogene 1998, 16, 3087–3095.
- Kao, S.C.; Fulham, M.; Wong, K.; Cooper, W.; Brahmbhatt, H.; MacDiarmid, J.; Pattinson, S.; Sagong, J.O.; Huynh, Y.; Leslie, F.; et al. A Significant Metabolic and Radiological Response after a Novel Targeted MicroRNA-based Treatment Approach in Malignant Pleural Mesothelioma. Am. J. Respir. Crit. Care Med. 2015, 191, 1467–1469.
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396.
- Reid, G.; Johnson, T.G.; Van Zandwijk, N. Manipulating microRNAs for the Treatment of Malignant Pleural Mesothelioma: Past, Present and Future. Front. Oncol. 2020, 10, 105.
- Rump, A.; Morikawa, Y.; Tanaka, M.; Minami, S.; Umesaki, N.; Takeuchi, M.; Miyajima, A. Binding of Ovarian Cancer Antigen CA125/MUC16 to Mesothelin Mediates Cell Adhesion. J. Biol. Chem. 2004, 279, 9190–9198.
- Gubbels, J.A.; Belisle, J.; Onda, M.; Rancourt, C.; Migneault, M.; Ho, M.; Bera, T.K.; Connor, J.; Sathyanarayana, B.K.; Lee, B.; et al. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol. Cancer 2006, 5, 50.
- Hassan, R.; Ebel, W.; Routhier, E.L.; Patel, R.; Kline, J.B.; Zhang, J.; Chao, Q.; Jacob, S.; Turchin, H.; Gibbs, L.; et al. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 2007, 7, 1–10.
- Hassan, R.; Schweizer, C.; Lu, K.F.; Schuler, B.; Remaley, A.T.; Weil, S.C.; Pastan, I. Inhibition of mesothelin–CA-125 interaction in patients with mesothelioma by the anti-mesothelin monoclonal antibody MORAb-009: Implications for cancer therapy. Lung Cancer 2010, 68, 455–459.
- Hassan, R.; Lerner, M.R.; Benbrook, R.; Lightfoot, S.A.; Brackett, D.J.; Wang, Q.-C.; Pastan, I. Antitumor activity of SS(dsFv)PE38 and SS1(dsFv)PE38, recombinant antimesothelin immunotoxins against human gynecologic cancers grown in organotypic culture in vitro. Clin. Cancer Res. 2002, 8, 3520–3526.
- Li, Q.; Verschraegen, C.F.; Mendoza, J.; Hassan, R. Cytotoxic activity of the recombinant anti-mesothelin immunotoxin, SS1(dsFv)PE38, towards tumor cell lines established from ascites of patients with peritoneal mesotheliomas. Anticancer. Res. 2004, 24, 1327–1336.
- Hollevoet, K.; Mason-Osann, E.; Liu, X.-F.; Imhof-Jung, S.; Niederfellner, G.; Pastan, I. In Vitro and In Vivo Activity of the Low-Immunogenic Antimesothelin Immunotoxin RG7787 in Pancreatic Cancer. Mol. Cancer Ther. 2014, 13, 2040–2049.
- Alewine, C.; Xiang, L.; Yamori, T.; Niederfellner, G.; Bosslet, K.; Pastan, I. Efficacy of RG7787, a Next-Generation Mesothelin-Targeted Immunotoxin, against Triple-Negative Breast and Gastric Cancers. Mol. Cancer Ther. 2014, 13, 2653–2661.
- Golfier, S.; Kopitz, C.; Kahnert, A.; Heisler, I.; Schatz, C.A.; Stelte-Ludwig, B.; Mayer-Bartschmid, A.; Unterschemmann, K.; Bruder, S.; Linden, L.; et al. Anetumab Ravtansine: A Novel Mesothelin-Targeting Antibody–Drug Conjugate Cures Tumors with Heterogeneous Target Expression Favored by Bystander Effect. Mol. Cancer Ther. 2014, 13, 1537–1548.
- Scales, S.J.; Gupta, N.; Pacheco, G.; Firestein, R.; French, D.M.; Koeppen, H.; Rangell, L.; Barry-Hamilton, V.; Luis, E.; Chuh, J.; et al. An Antimesothelin-Monomethyl Auristatin E Conjugate with Potent Antitumor Activity in Ovarian, Pancreatic, and Mesothelioma Models. Mol. Cancer Ther. 2014, 13, 2630–2640.
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and Survival with GVAX Pancreas Prime and Listeria Monocytogenes–Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1325–1333.
- Hassan, R.; Alley, E.; Kindler, H.; Antonia, S.; Jahan, T.; Honarmand, S.; Nair, N.; Whiting, C.C.; Enstrom, A.; Lemmens, E.; et al. Clinical Response of Live-Attenuated, Listeria monocytogenes Expressing Mesothelin (CRS-207) with Chemotherapy in Patients with Malignant Pleural Mesothelioma. Clin. Cancer Res. 2019, 25, 5787–5798.
- Husson, A.; Brasse-Lagnel, C.; Fairand, A.; Renouf, S.; Lavoinne, A. Argininosuccinate synthetase from the urea cycle to the citrulline-NO cycle. JBIC J. Biol. Inorg. Chem. 2003, 270, 1887–1899.
- Mori, M.; Gotoh, T. Regulation of Nitric Oxide Production by Arginine Metabolic Enzymes. Biochem. Biophys. Res. Commun. 2000, 275, 715–719.
- Wallace, H.M.; Fraser, A.V.; Hughes, A. A perspective of polyamine metabolism. Biochem. J. 2003, 376, 1–14.
- Szlosarek, P.W.; Klabatsa, A.; Pallaska, A.; Sheaff, M.; Smith, P.; Crook, T.; Grimshaw, M.J.; Steele, J.P.; Rudd, R.M.; Balkwill, F.R.; et al. In vivo Loss of Expression of Argininosuccinate Synthetase in Malignant Pleural Mesothelioma Is a Biomarker for Susceptibility to Arginine Depletion. Clin. Cancer Res. 2006, 12, 7126–7131.
- Szlosarek, P.W.; Steele, J.P.; Nolan, L.; Gilligan, D.; Taylor, P.; Spicer, J.; Lind, M.; Mitra, S.; Shamash, J.; Phillips, M.M.; et al. Arginine Deprivation with Pegylated Arginine Deiminase in Patients with Argininosuccinate Synthetase 1-Deficient Malignant Pleural Mesothelioma: A Randomized Clinical Trial. JAMA Oncol. 2017, 3, 58–66.
- Beddowes, E.; Spicer, J.; Chan, P.Y.; Khadeir, R.; Corbacho, J.G.; Repana, D.; Steele, J.P.; Schmid, P.; Szyszko, T.; Cook, G.; et al. Phase 1 Dose-Escalation Study of Pegylated Arginine Deiminase, Cisplatin, and Pemetrexed in Patients with Argininosuccinate Synthetase 1-Deficient Thoracic Cancers. J. Clin. Oncol. 2017, 35, 1778–1785.
- Alley, E.W.; Lopez, J.; Santoro, A.; Morosky, A.; Saraf, S.; Piperdi, B.; van Brummelen, E. Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): Preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2017, 18, 623–630.
- Danson, S.J.; Conner, J.; Edwards, J.G.; Blyth, K.G.; Fisher, P.M.; Muthana, M.; Salawu, A.; Taylor, F.; Hodgkinson, E.; Joyce, P.; et al. Oncolytic herpesvirus therapy for mesothelioma—A phase I/IIa trial of intrapleural administration of HSV1716. Lung Cancer 2020, 150, 145–151.
- Robinson, B.; Robinson, C.; Lake, R. Localised spontaneous regression in mesothelioma—Possible immunological mechanism. Lung Cancer 2001, 32, 197–201.
- Anraku, M.; Cunningham, K.S.; Yun, Z.; Tsao, M.; Zhang, L.; Keshavjee, S.; Johnston, M.R.; de Perrot, M. Impact of tumor-infiltrating T cells on survival in patients with malignant pleural mesothelioma. J. Thorac. Cardiovasc. Surg. 2008, 135, 823–829.
- Leigh, R.A.; Webster, I. Lymphocytic infiltration of pleural mesothelioma and its significance for survival. S. Afr. Med. J. 1982, 61, 1007–1009.
- Balar, A.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564.
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264.
- Rigo, V.; Emionite, L.; Daga, A.; Astigiano, S.; Corrias, M.V.; Quintarelli, C.; Locatelli, F.; Ferrini, S.; Croce, M. Combined immunotherapy with anti-PDL-1/PD-1 and anti-CD4 antibodies cures syngeneic disseminated neuroblastoma. Sci. Rep. 2017, 7, 14049.
- Currie, A.J.; Prosser, A.; McDonnell, A.; Cleaver, A.L.; Robinson, B.W.S.; Freeman, G.J.; van der Most, R.G. Dual Control of Antitumor CD8 T Cells through the Programmed Death-1/Programmed Death-Ligand 1 Pathway and Immunosuppressive CD4 T Cells: Regulation and Counterregulation. J. Immunol. 2009, 183, 7898–7908.
- Maio, M.; Scherpereel, A.; Calabro’, L.; Aerts, J.; Perez, S.C.; Bearz, A.; Nackaerts, K.; Fennell, D.A.; Kowalski, D.; Tsao, A.S.; et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): A multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. 2017, 18, 1261–1273.
- Cantini, L.; Hassan, R.; Sterman, D.H.; Aerts, J.G.J.V. Emerging Treatments for Malignant Pleural Mesothelioma: Where Are We Heading? Front Oncol. 2020, 10, 343.
- Tallón de Lara, P.; Cecconi, V.; Hiltbrunner, S.; Yagita, H.; Friess, M.; Bode, B.; Opitz, I.; Vrugt, B.; Weder, W.; Stolzmann, P.; et al. Gemcitabine Synergizes with Immune Checkpoint Inhibitors and Overcomes Resistance in a Preclinical Model and Mesothelioma Patients. Clin. Cancer Res. 2018, 24, 6345–6354.
- Aston, W.J.; Fisher, S.A.; Khong, A.; Mok, C.; Nowak, A.K.; Lake, R.A.; Lesterhuis, W.J. Combining chemotherapy and checkpoint blockade in thoracic cancer: How to proceed? Lung Cancer Manag. 2014, 3, 443–457.
- Wu, L.; Wu, M.O.; De La Maza, L.; Yun, Z.; Yu, J.; Zhao, Y.; Cho, J.; De Perrot, M. Targeting the inhibitory receptor CTLA-4 on T cells increased abscopal effects in murine mesothelioma model. Oncotarget 2015, 6, 12468–12480.
- Kao, S.C.; Cheng, Y.Y.; Williams, M.; Kirschner, M.B.; Madore, J.; Lum, T.; Sarun, K.H.; Linton, A.; McCaughan, B.; Klebe, S.; et al. Tumor Suppressor microRNAs Contribute to the Regulation of PD-L1 Expression in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2017, 12, 1421–1433.
- Klampatsa, A.; Achkova, D.Y.; Davies, D.M.; Parente-Pereira, A.C.; Woodman, N.; Rosekilly, J.; Osborne, G.; Thayaparan, T.; Bille, A.; Sheaf, M.; et al. Intracavitary ‘T4 immunotherapy’ of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017, 393, 52–59.
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120.
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.-C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.; June, C.H.; Albelda, S.M. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T Cells Expressing a Mesothelin-Specific Chimeric Antibody Receptor. Clin. Cancer Res. 2011, 17, 4719–4730.
- Zeltsman, M.; Dozier, J.; McGee, E.; Ngai, D.; Adusumilli, P.S. CAR T-cell therapy for lung cancer and malignant pleural mesothelioma. Transl. Res. 2017, 187, 1–10.
- Tay, B.; Wright, Q.; Ladwa, R.; Perry, C.; Leggatt, G.; Simpson, F.; Wells, J.; Panizza, B.; Frazer, I.; Cruz, J. Evolution of Cancer Vaccines—Challenges, Achievements, and Future Directions. Vaccines 2021, 9, 535.
- Creaney, J.; Ma, S.; Sneddon, S.A.; Tourigny, M.R.; Dick, I.M.; Leon, J.S.; Khong, A.; Fisher, S.A.; Lake, R.A.; Lesterhuis, W.J.; et al. Strong spontaneous tumor neoantigen responses induced by a natural human carcinogen. Oncoimmunology 2015, 4, e1011492.
- Sneddon, S.; Rive, C.; Ma, S.; Dick, I.M.; Allcock, R.J.N.; Brown, S.D.; Holt, R.; Watson, M.; Leary, S.; Lee, Y.C.G.; et al. Identification of a CD8+ T-cell response to a predicted neoantigen in malignant mesothelioma. OncoImmunology 2019, 9, 1684713.
- Pease, D.F.; Kratzke, R.A. Oncolytic Viral Therapy for Mesothelioma. Front. Oncol. 2017, 7, 179.
- Di Somma, S.; Iannuzzi, C.A.; Passaro, C.; Forte, I.M.; Iannone, R.; Gigantino, V.; Indovina, P.; Botti, G.; Giordano, A.; Formisano, P.; et al. The Oncolytic Virus dl922-947 Triggers Immunogenic Cell Death in Mesothelioma and Reduces Xenograft Growth. Front. Oncol. 2019, 9, 564.
- Li, H.; Peng, K.-W.; Dingli, D.; Kratzke, R.A.; Russell, S.J. Oncolytic measles viruses encoding interferon β and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010, 17, 550–558.
- Kucharczuk, J.C.; Randazzo, B.; Chang, M.Y.; Amin, K.M.; Elshami, A.A.; Sterman, D.; Rizk, N.P.; Molnar-Kimber, K.L.; Brown, S.M.; MacLean, A.R.; et al. Use of a “replication-restricted” herpes virus to treat experimental human malignant mesothelioma. Cancer Res. 1997, 57, 466–471.
- Danson, S.; Woll, P.; Edwards, J.; Blyth, K.; Fisher, P.; Roman, J.; Simpson, K.; Spavin, R.; Learmonth, K.; Conner, J. 366PD—Oncolytic herpesvirus therapy for mesothelioma: A phase I/IIa trial of intrapleural administration of HSV1716 (NCT01721018). Ann. Oncol. 2017, 28, v122.
- Friedlander, P.L.; Delaune, C.L.; Abadie, J.M.; Toups, M.; Lacour, J.; Marrero, L.; Zhong, Q.; Kolls, J.K. Efficacy of CD40 Ligand Gene Therapy in Malignant Mesothelioma. Am. J. Respir. Cell Mol. Biol. 2003, 29, 321–330.
- Belin, L.J.; Ady, J.W.; Lewis, C.; Marano, D.; Gholami, S.; Mojica, K.; Eveno, C.; Longo, V.; Zanzonico, P.B.; Chen, N.G.; et al. An oncolytic vaccinia virus expressing the human sodium iodine symporter prolongs survival and facilitates SPECT/CT imaging in an orthotopic model of malignant pleural mesothelioma. Surgery 2013, 154, 486–495.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
834
Revisions:
2 times
(View History)
Update Date:
12 Oct 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No