 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Javier Leon | + 1860 word(s) | 1860 | 2021-09-22 11:41:17 | | | |
| 2 | Vicky Zhou | -3 word(s) | 1857 | 2021-10-02 03:39:48 | | |
Video Upload Options
MNT (MAX’s Next Tango) is a crucial modulator of MYC, controls several cellular functions, and is activated in most human cancers. It is the largest, most divergent, and most ubiquitously expressed protein of the MXD family. MNT was first described as a MYC antagonist and tumor suppressor. Indeed, 10% of human tumors present deletions of one MNT allele. However, some reports show that MNT functions in cooperation with MYC by maintaining cell proliferation, promoting tumor cell survival, and supporting MYC-driven tumorigenesis in cellular and animal models. Although MAX was originally considered MNT’s obligate partner, it is also known that heterodmerize with MLX. Recent findings demonstrate that MNT can also work independently, as MNT forms homodimers and interacts with proteins both outside and inside of the proximal MYC network, as REL. These complexes are involved in a wide array of cellular processes, from transcriptional repression via SIN3 to the modulation of metabolism through MLX as well as immunity and apoptosis via REL.
1. MNT discovery and structure
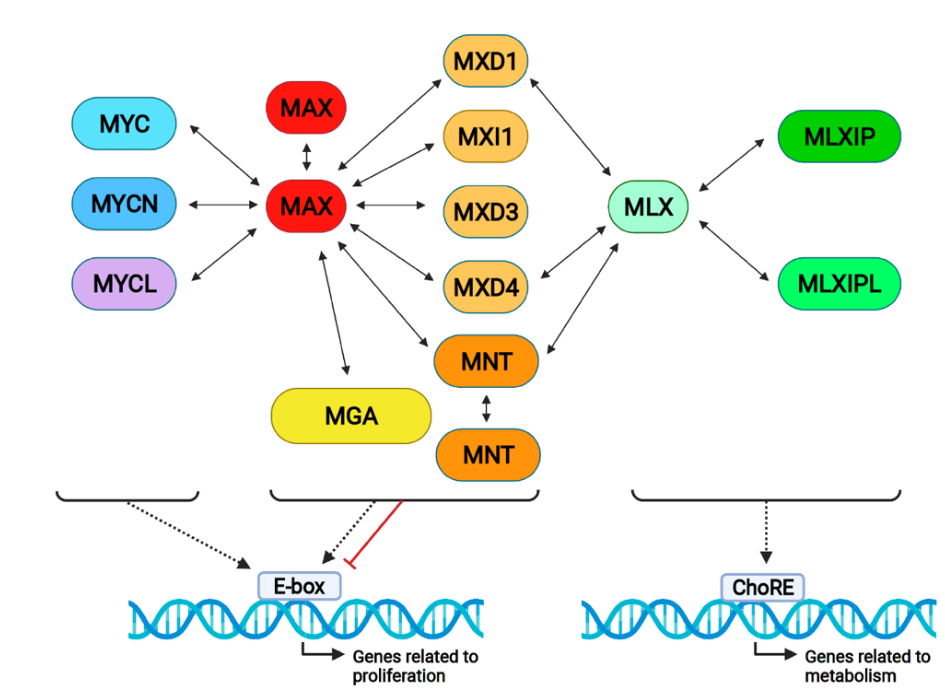 Figure 1. The proximal MYC network and its interactions. MYC proteins form dimers with MAX and bind DNA on E-boxes, generally activating transcription. All MXD proteins bind to MAX and MXD1, and MXD4 and MNT also bind to MLX, activating or repressing gene transcription. MLX forms complexes with MLXIP (MONDOA) and MLXIPL (MONDOB), binding on ChoRE (carbohydrate response elements) and regulating metabolic genes. Notably, MAX and MNT form homodimers, as
Figure 1. The proximal MYC network and its interactions. MYC proteins form dimers with MAX and bind DNA on E-boxes, generally activating transcription. All MXD proteins bind to MAX and MXD1, and MXD4 and MNT also bind to MLX, activating or repressing gene transcription. MLX forms complexes with MLXIP (MONDOA) and MLXIPL (MONDOB), binding on ChoRE (carbohydrate response elements) and regulating metabolic genes. Notably, MAX and MNT form homodimers, asrepresented in the Figure.
| MNT | MXD1 | MXI1 (MXD2) | MXD3 | MXD4 | MYC | References | |
|---|---|---|---|---|---|---|---|
| Protein size | 582 aa | 221 aa | 228 aa | 206 aa | 209 aa | 439 aa | Uniprot database |
| P-rich sequences | Yes | No | No | No | No | Yes | [2] |
| Phenotype of KO mice | Perinatally lethal/Craniofacial abnormalities | Viable/Increased immature granulocyte progenitors | Viable/Hyperplasia | Viable/Enhanced sensitivity to apoptotic stimuli | ND | Embryonic lethal E9.5–E10.5 | [11][12][13][14] |
| Proximal MYC Network interactors | MNT, MAX, MLX | MAX, MLX | MAX | MAX | MAX, MLX | MAX | Reviewed in [15] |
| Expression in cells | Quiescent and proliferating | Quiescent | Quiescent and proliferating * | Proliferating (S-phase) | Quiescent | Proliferating | Reviewed in [15] |
| Pan-cancer copy number alterations (%) | DEL 10 AMP 3 MUT 1 |
DEL 2 AMP 6 MUT < 0.5 |
DEL 8 AMP 4 MUT < 0.5 |
DEL 7 AMP 8 MUT < 0.5 |
DEL 6 AMP 5 MUT < 0.5 |
DEL 2 AMP 21 MUT 1 |
[16] |
| Involvement in human cancer | DEL in CTCL-SS and ALL. Reduced expression in MB | None or weak | None or weak | None or weak | None or weak | Strongly, 70% of tumors have deregulation | [17][18][19][20][21][22] |
2. MNT Alterations in Cancer
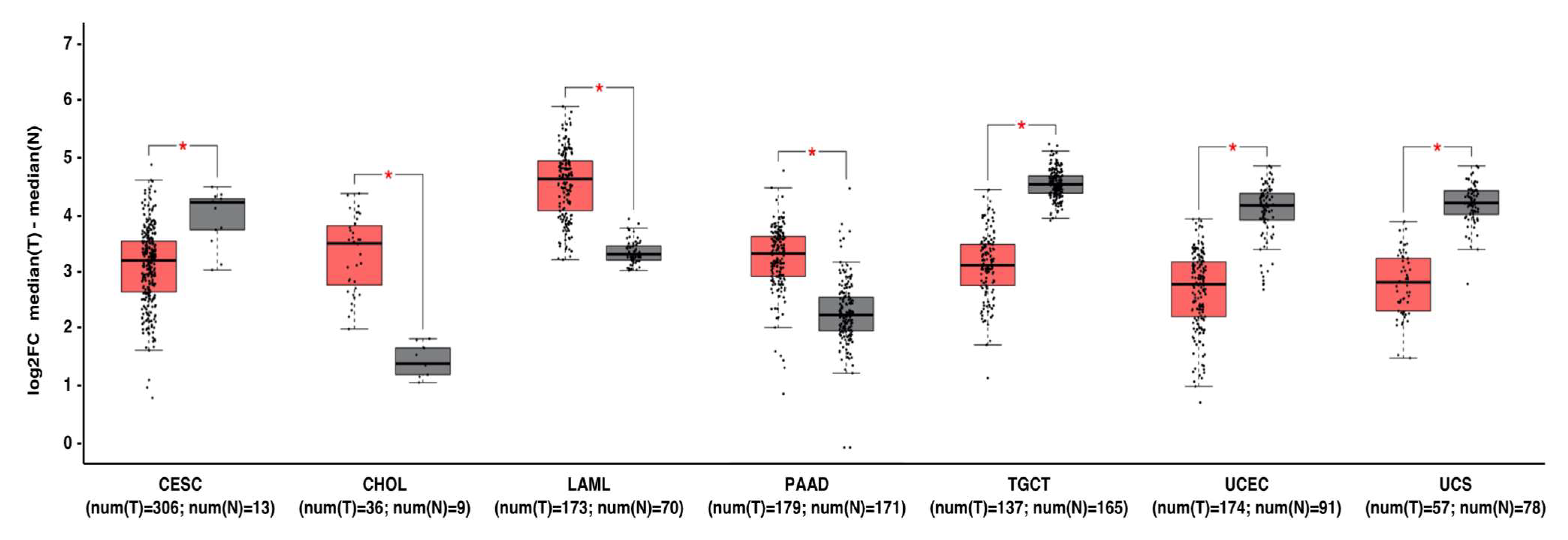
Figure 2. MNT can act as a tumor suppressor gene or oncogene depending on the tumor type. Gene expression profiling interactive analysis (GEPIA, http://gepia.cancer-pku.cn/) was performed to validate MNT mRNA expression in selected cancer type samples (in red) vs. normal samples (in grey). The expression data were first log2(TPM+1) transformed for differential analysis, and the log2FC was defined as median (tumor)–median (normal). Data are represented as mean ± SD (* p < 0.01). Abbreviations: CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, Cholangiocarcinoma; LAML, acute myeloid leukemia; PAAD, pancreatic adenocarcinoma; TGCT, testicular germ cell tumors; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma.
3. Concluding Remarks
References
- Hurlin, P.J.; Quéva, C.; Eisenman, R.N. Mnt, a novel Max-interacting protein is coexpressed with Myc in proliferating cells and mediates repression at Myc binding sites. Genes Dev. 1997, 11, 44–58.
- Meroni, G.; Reymond, A.; Alcalay, M.; Borsani, G.; Tanigami, A.; Tonlorenzi, R.; Lo Nigro, C.; Messali, S.; Zollo, M.; Ledbetter, D.H.; et al. Rox, a novel bHLHZip protein expressed in quiescent cells that heterodimerizes with Max, binds a non-canonical E box and acts as a transcriptional repressor. EMBO J. 1997, 16, 2892–2906.
- Ayer, D.E.; Lawrence, Q.A.; Eisenman, R.N. Mad-max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell 1995, 80, 767–776.
- Grzenda, A.; Lomberk, G.; Zhang, J.S.; Urrutia, R. Sin3: Master scaffold and transcriptional corepressor. Biochim. Biophys. Acta Gene Regul. Mech. 2009, 1789, 443–450.
- Raffeiner, P.; Hart, J.R.; García-Caballero, D.; Bar-Peled, L.; Weinberg, M.S.; Vogt, P.K. An MXD1-derived repressor peptide identifies noncoding mediators of MYC-driven cell proliferation. Proc. Natl. Acad. Sci. USA 2020, 117, 6571–6579.
- Hurlin, P.J.; Steingrimsson, E.; Copeland, N.G.; Jenkins, N.A.; Eisenman, R.N. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA- binding motif. EMBO J. 1999, 18, 7019–7028.
- Rikin, A.; Evans, T. The Tbx/bHlH Transcription Factor mga regulates gata4 and Organogenesis. Dev. Dyn. 2010, 239, 535–547.
- Ogawa, H.; Ishiguro, K.; Gaubatz, S.; Livingston, D.M.; Nakatani, Y. A Complex with Chromatin Modifiers That Occupies E2F- and Myc-Responsive Genes in G0 Cells. Science 2002, 296, 1132–1136.
- Mathsyaraja, H.; Catchpole, J.; Eastwood, E.; Babaeva, E.; Geuenich, M.; Cheng, P.F.; Freie, B.; Ayers, J.; Yu, M.; Wu, N.; et al. Loss of MGA mediated Polycomb repression promotes tumor progression and invasiveness. Elife 2021, 28, e64212.
- Yang, G.; Hurlin, P.J. MNT and Emerging Concepts of MNT-MYC Antagonism. Genes 2017, 8, 83.
- Toyo-oka, K.; Hirotsune, S.; Gambello, M.J.; Zhou, Z.; Olson, L.; Rosenfeld, M.G.; Eisenman, R.; Hurlin, P.; Wynshaw-Boris, A. Loss of the Max-interacting protein Mnt in mice results in decreased viability, defective embryonic growth and craniofacial defects: Relevance to Miller-Dieker syndrome. Hum. Mol. Genet. 2004, 13, 1057–1067.
- Foley, K.P.; McArthur, G.A.; Quéva, C.; Hurlin, P.J.; Soriano, P.; Eisenman, R.N. Targeted disruption of the MYC antagonist MAD1 inhibits cell cycle exit during granulocyte differentiation. EMBO J. 1998, 17, 774–785.
- Schreiber-Agus, N.; Meng, Y.; Hoang, T.; Hou, H.; Chen, K.; Greenberg, R.; Cordon-Cardo, C.; Lee, H.-W.; DePinho, R.A. Role of Mxi1 in ageing organ systems and the regulation of normal and neoplastic growth. Nature 1998, 393, 483–487.
- Queva, C.; McArthur, G.A.; Iritani, B.M.; Eisenman, R.N. Targeted Deletion of the S-Phase-Specific Myc Antagonist Mad3 Sensitizes Neuronal and Lymphoid Cells to radiation-induced apoptosis. Mol. Cell. Biol. 2001, 21, 703–712.
- Diolaiti, D.; McFerrin, L.; Carroll, P.A.; Eisenman, R.N. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta 2015, 1849, 484–500.
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2.
- Boonk, S.E.; Zoutman, W.H.; Marie-Cardine, A.; van der Fits, L.; Out-Luiting, J.J.; Mitchell, T.J.; Tosi, I.; Morris, S.L.; Moriarty, B.; Booken, N.; et al. Evaluation of Immunophenotypic and Molecular Biomarkers for Sézary Syndrome Using Standard Operating Procedures: A Multicenter Study of 59 Patients. J. Investig. Dermatol. 2016, 136, 1364–1372.
- Van Doorn, R.; Dijkman, R.; Vermeer, M.H.; Out-Luiting, J.J.; Van Der Raaij-Helmer, E.M.H.; Willemze, R.; Tensen, C.P. Aberrant expression of the tyrosine kinase receptor EphA4 and the transcription factor twist in Sézary syndrome identified by gene expression analysis. Cancer Res. 2004, 64, 5578–5586.
- Vermeer, M.H.; Van Doorn, R.; Dijkman, R.; Mao, X.; Whittaker, S.; Van Voorst Vader, P.C.; Gerritsen, M.J.P.; Geerts, M.L.; Gellrich, S.; Söderberg, O.; et al. Novel and highly recurrent chromosomal alterations in Sézary Syndrome. Cancer Res. 2008, 68, 2689–2698.
- Guo, X.; Pan, L.; Zhang, X.; Suo, X.; Niu, Z.; Zhang, J.; Wang, F.; Dong, Z.; Da, W.; Ohno, R. Expression and mutation analysis of genes that encode the Myc antagonists Mad1, Mxi1 and Rox in acute leukaemia. Leuk. Lymphoma 2007, 48, 1200–1207.
- Cvekl, A.; Zavadil, J.; Birshtein, B.K.; Grotzer, M.A.; Cvekl, A. Analysis of transcripts from 17p13.3 in medulloblastoma suggests ROX/MNT as a potential tumour suppressor gene. Eur. J. Cancer 2004, 40, 2525–2532.
- Wahlström, T.; Henriksson, M. Mnt Takes Control as Key Regulator of the Myc/Max/Mxd Network. Adv. Cancer Res. 2007, 97, 61–80.
- Loo, L.W.M.; Secombe, J.; Little, J.T.; Leni-Sue Carlos, L.-S.; Yost, C.; Cheng, P.-F.; Flynn, E.M.; Edgar, B.A.; Eisenman, R.N. The Transcriptional Repressor dMnt Is a Regulator of Growth in Drosophila melanogaster. Mol. Cell. Biol. 2005, 25, 7078–7091.
- Orian, A.; van Steensel, B.; Delrow, J.; Bussemaker, H.J.; Li, L.; Sawado, T.; Williams, E.; Loo, L.W.M.; Cowley, S.M.; Yost, C.; et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003, 17, 1101–1114.
- McFerrin, L.G.; Atchley, W.R. Evolution of the Max and Mlx networks in animals. Genome Biol. Evol. 2011, 3, 915–937.
- Johnson, D.W.; Llop, J.R.; Farrell, S.F.; Yuan, J.; Stolzenburg, L.R.; Samuelson, A.V. The Caenorhabditis elegans Myc-Mondo/Mad Complexes Integrate Diverse Longevity Signals. PLoS Genet. 2014, 10, e1004278.
- Hurlin, P.J.; Zhou, Z.; Toyo-oka, K.; Ota, S.; Walker, W.L.; Hirotsune, S.; Wynshaw-boris, A. Deletion of Mnt leads to disrupted cell cycle control and tumorigenesis. EMBO J. 2003, 22, 4584–4596.
- Schreiber-Agus, N.; Chin, L.; Chen, K.; Torres, R.; Rao, G.; Guida, P.; Skoultchi, A.; Depinho, R.A. An Amino-Terminal Domain of Mxi1 Mediates Anti-Myc Oncogenic Activity and Interacts with a Homolog of the Yeast Transcriptional Repressor SIN3. Cell 1995, 80, 777–786.
- Dezfouli, S.; Bakke, A.; Huang, J.; Wynshaw-Boris, A.; Hurlin, P.J. Inflammatory Disease and Lymphomagenesis Caused by Deletion of the Myc Antagonist Mnt in T Cells. Mol. Cell. Biol. 2006, 26, 2080–2092.
- Toyo-oka, K.; Bowen, T.J.; Hirotsune, S.; Li, Z.; Jain, S.; Ota, S.; Lozach, L.E.; Bassett, I.G.; Lozach, J.; Rosenfeld, M.G.; et al. Mnt-Deficient Mammary Glands Exhibit Impaired Involution and Tumors with Characteristics of Myc Overexpression. Cancer Res. 2006, 66, 5565–6894.
- Edelmann, J.; Holzmann, K.; Miller, F.; Winkler, D.; Buhler, A.; Zenz, T.; Bullinger, L.; Kuhn, M.W.M.; Gerhardinger, A.; Bloehdorn, J.; et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood 2012, 6, 4783–4794.
- Lo Nigro, C.; Venesio, T.; Reymond, A.; Meroni, G.; Alberici, P.; Cainarca, S.; Enrico, F.; Stack, M.; Ledbetter, D.H.; Liscia, D.S.; et al. The Human ROX Gene: Genomic Structure and Mutation Analysis in Human Breast Tumors. Genomics 1998, 282, 275–282.
- Sommer, A.; Waha, A.; Tonn, J.; Sörensen, N.; Hurlin, P.J.; Eisenman, R.N.; Lüscher, B.; Pietsch, T. Analysis of the MAX-binding protein MNT in human medulloblastomas. Int. J. Cancer 1999, 82, 810–816.
- Phillips, N.; Ziegler, M.; Saha, B.; Xynos, F. Allelic loss on chromosome 17 in human ovarian cancer. Int. J. Cancer 1993, 54, 85–91.
- Saxena, A.; Clark, W.C.; Robertson, J.T.; Ikejiri, B.; Oldfield, E.H.; Ali, I.U. Erratum: Evidence for the involvement of a potential second tumor suppressor gene on chromosome 17 distinct from p53 in malignant astrocytomas (Cancer Research (December 1, 1992) (6716–6721)). Cancer Res. 1993, 53, 1472.
- Williamson, M.P.; Elder, P.A.; Knowles, M.A. The spectrum of TP53 mutations in bladder carcinoma. Genes Chromosom. Cancer 1994, 9, 108–118.
- Andreassen, Å.; Oyjord, T.; Hovig, E.; Holm, R.; Florenes, V.A.; Nesland, J.M.; Myklebost, O.; Hoie, J.; Bruland, S.; Borresen, A.-L.; et al. p53 Abnormalities in Different Subtypes of Human Sarcomas. Cancer Res. 1993, 53, 468–471.
- Sheiness, D.; Fanshier, L.; Bishop, J.M. Identification of nucleotide sequences which may encode the oncogenic capacity of avian retrovirus MC29. J. Virol. 1978, 28, 600–610.
- Sheiness, D.; Bishop, J.M. DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J. Virol. 1979, 31, 514–521.
- Duesberg, P.H.; Bister, K.; Vogt, P.K. The RNA of avian acute leukemia virus MC29. Proc. Natl. Acad. Sci. USA 1977, 74, 4320–4324.
- Massó-Vallés, D.; Soucek, L. Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells 2020, 9, 883.
- Nilsson, J.A.; Maclean, K.H.; Keller, U.B.; Pendeville, H.; Baudino, T.A.; Cleveland, J.L. Mnt loss triggers Myc transcription targets, proliferation, apoptosis, and transformation. Mol. Cell. Biol. 2004, 24, 1560–1569.
- Nguyen, H.V.; Vandenberg, C.J.; Ng, A.P.; Robati, M.R.; Anstee, N.S.; Rimes, J.; Hawkins, E.D.; Cory, S. Development and survival of MYC-driven lymphomas require the MYC antagonist MNT to curb MYC-induced apoptosis. Blood 2020, 135, 1019–1031.
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006, 16, 318–330.
- Castell, A.; Yan, Q.; Fawkner, K.; Hydbring, P.; Zhang, F.; Verschut, V.; Franco, M.; Zakaria, S.M.; Bazzar, W.; Goodwin, J.; et al. A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 2018, 8, 10064.
- Demma, M.J.; Hohn, M.J.; Sun, A.; Mapelli, C.; Hall, B.; Walji, A.; O’Neil, J. Inhibition of Myc transcriptional activity by a mini-protein based upon Mxd1. FEBS Lett. 2020, 594, 1467–1476.
- Demma, M.J.; Mapelli, C.; Sun, A.; Bodea, S.; Ruprecht, B.; Javaid, S.; Wiswell, D.; Muise, E.; Chen, S.; Zelina, J.; et al. Omomyc Reveals New Mechanisms To Inhibit the MYC Oncogene. Mol. Cell. Biol. 2019, 39, 1–27.


