 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carmela Conte | + 3714 word(s) | 3714 | 2021-09-28 13:07:16 | | | |
| 2 | Catherine Yang | -1921 word(s) | 1793 | 2021-09-29 09:52:19 | | |
Video Upload Options
Toll-like receptors (TLRs) are type I transmembrane glycoproteins belonging to the complex pattern recognition receptors (PRRs) expressed in immune and non-immune cells, including neurons and glia, which are involved in the regulation of innate immune and inflammatory responses. The TLR family comprises 11 members (TLR1–TLR11) in human and 12 (TLR1–TLR9, TLR11–TLR13) in mouse. TLR4 is one of the best characterized TLRs that surveys for the presence of structural motifs in a wide array of invading microorganisms, named pathogen-associated molecular patterns (PAMPs), and endogenous damage or danger molecular patterns (DAMPs), also known as alarmins, released by damaged cells and injured tissues or derived from apoptotic and necrotic cells. Activation of TLR4 induces the downstream start of inflammasome pathways which results in the release of a plethora of pro-inflammatory cytokines, Type I interferons (IFNs) and other inflammatory mediators.
1. Introduction
TLR4 can recognize a wide array of DAMPs or PAMPs including invading bacteria, viruses, parasites, and cell debris [1][2]. In particular, TLR4 is highly specialized in sensing lipopolysaccharide (LPS) from Gram-negative. The formation of the complex TLR4-myelod differentiation 2 (MD2) on the cell surface, as well as the presence of LPS-binding proteins and cluster of differentiation 14 (CD14), is required for the recognition of ligands and the activation of intracellular pathways. The dimerization of TLR4-MD2 induces conformational changes that endorse the recruitment of adaptor proteins containing Toll/interleukin-1 receptor-like (TIR) domains. This is essential to start two possible intracellular signaling pathways: the MyD88-dependent pathway that leads to the release of proinflammatory cytokines, and the MyD88-independent pathway that causes the production of the type I interferons [3]. The engagement of MyD88 and MyD88-adaptor-like (MAL) protein induces a cascade of downstream events including the phosphorylation of IL-1R-associated kinases (IRAKs), the association of TNF-receptor-associated factor 6 (TRAF6), and the activation of transforming growth factor β-activated kinase 1 (TAK1). Many other adaptor proteins are involved and include TAK1-binding protein 2 (TAB2) and TAK1-binding protein 3 (TAB3). TAK1, in turn, activates two different kinase types: the mitogen-activated protein kinases (MAPKs), which comprise JUN N-terminal kinase (JNK), p38, extracellular signal-regulated kinases (ERK1/2), and the IkB kinase complex (IKK). The final response is the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and activator protein-1 (AP1) and the abundant secretion of proinflammatory cytokines [4] Instead, the activation of the MyD88-independent pathway is started by internalization of the TLR4-MD2 complex by CD14 and the recruitment/activation of adaptor proteins TRIF, TRIF-related adaptor molecule (TRAM), and TRAF3 followed by IFN regulatory factor 3 (IRF3) nuclear translocations. These events culminate with the production of type I IFNs [5]. Sustained activation of these signals attracts macrophages, natural killer, and other cells and may result in the toxic accumulation of ROS and RNS and cell death [6]
2. The Role of TLR4 in neuroinflammation and Parkinson’s disease
According to the World Health Organization (WHO), Parkinson’s disease (PD) is the second most common neurodegenerative disease in the world, after Alzheimer’s disease (AD), and one of the most common causes of neurological disability with a high social impact [7]. The prevalence of PD is about 10 million people worldwide, and it is estimated that there will be about 13 million people with PD by 2040 [8]. PD is a chronic, progressive, age-related neurodegenerative disease clinically characterized by motor symptoms, such as bradykinesia, rigidity, tremor at rest, slow movements, and postural instability [9]. Motor symptoms are the consequence of the progressive loss of neuromelanin-containing neurons, especially dopaminergic neurons (DA) in the substantia nigra pars compacta (SNpc) and dopamine depletion in the striatum, as confirmed by post-mortem study of clinically diagnosed PD patients [10][11]. The pathological hallmark of PD is represented by the accumulation of cytoplasmic inclusions such as Lewy bodies (LB) and Lewy neurites rich in α-synuclein (α-SYN) [12][13][14].
Neuroinflammation is a complex process able to activate the innate immune response in the central nervous system (CNS), associated with neurodegenerative diseases. For a long time, the CNS has been considered an immunologically privileged site because the lack of cells capable of presenting the antigen in the brain parenchyma, the absence of lymphatic drainage, and the presence of blood brain barrier (BBB). Over the last 30 years, this concept has been challenged, and many reports show active cooperation between the CNS and immune system despite the physical barrier [15][16]. A growing body of evidence indicates that neuroinflammation is involved in the pathogenesis of several neurodegenerative diseases including PD [17][18][19][20], Alzheimer’s disease (AD) [21], amyotrophic lateral sclerosis [22], and Huntington’s disease [23][24]. Neuroinflammation implicates the recruitment of immune and non-immune cells at the site of injury or infection, such as resident macrophages, i.e., microglia, aside from astrocytes, endothelial cells, infiltrating T-lymphocytes, α-SYN reactive T-lymphocytes, and major histocompatibility complex (MHC) class II-positive microglia [25][26][27][28]. Microglia are the most abundant populations in the brain parenchyma and represent the major cell type involved in neuroinflammation. However, sophisticated neuron–microglia–astrocyte crosstalk also plays a critical role in the immune response [28][29][30][31]. Although no standardized morphological classification exists, the mature brain microglial cells are generally quiescent or “resting” and exhibit a ramified shape with fine extending protrusions. They also are equipped with a wide range of receptors, comprising colony-stimulating factor 1 receptors that continuously scan the surrounding environment and control important functions such as synaptic activity, clearance of cell debris, and cerebral homeostasis [32][33][34][35]. Whether neuroinflammation is a consequence or a cause of nigral cell loss in PD is still unclear. Certainly, PD is known to impact the switch of microglia from the neuroprotective to the neurotoxic phenotype [36][37][38][39]. Sustained gliosis appears to be the prominent pathological feature of PD that is associated with loss of dopaminergic neurons in the SNpc [40][41][42] [Figure 1].
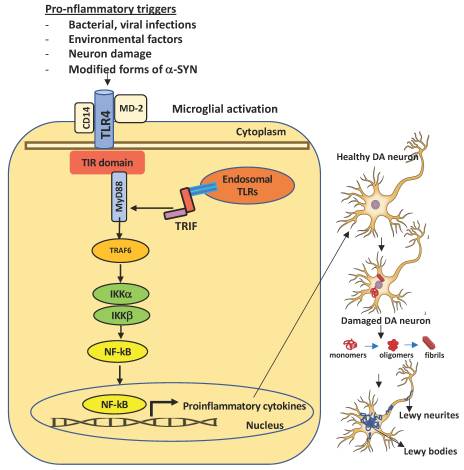
Figure 1. Schematic representation of TLR4 activation in microglia and consequent dopaminergic neuronal damage in the substantia nigra. A number of pro-inflammatory stimuli can promote the activation of the intracellular TLR4 signaling pathway in microglia. Upon brain injury, DAMPs or PAMPs released in the extracellular milieu via exocytosis from neighboring neurons are sensed by microglial TLR4 for degradation and clearance. If compromised or if prolonged, this process can lead to further inflammatory signaling in which the engagement of myeloid differentiating primary response gene 88 (MyD88) or TIR-domain containing adapter inducing interferon β (TRIF) provokes the activation of downstream signaling cascades that lead to the proteasomal degradation of kB inhibitors (IkB), release, and nuclear translocation of the NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells) transcription factor. The transcriptional activation of specific genes induces the release of proinflammatory cytokines, that in turn can cause neuronal damage, pathological modification of α-SYN (monomers, oligomers, and fibrils, truncated and phosphorylated forms), and aggregation in Lewy and neurites bodies—the pathological hallmarks of degenerating neurons in Parkinson’s disease. Endosomal TLRs sense bacterial or viral nucleic acids.
Therefore, immunological and inflammatory stimuli can promote phenotype switching between a highly ramified morphology and a large ameboid-like shape with short branches typical of activated microglia [43][44][45]. This switching triggers a complex cascade of events whose magnitude depends on the intensity and duration of the stimulation [46]. While the acute insults evoke protection of brain tissue, chronic stimulation causes a sustained inflammatory response which can cause the acquisition of a number of functions accompanied by the upregulation of a large number of receptor types, such as cytokine receptors, TLRs or cell adhesion molecules. Notably, the degeneration of the dopaminergic neurons can be caused not only by glial response but also by the loss of supporting role by resting microglia following switching to an activated status. Neuroinflammation in PD is also accompanied by microglia uptake and phagocytosis of neuromelanin released by degenerated DA neurons into the extracellular compartment that contribute to the acceleration of the neurodegenerative process. A number of studies suggest that TLRs, mainly TLR2 and 4 participate as promoters of immune/neuroinflammatory responses that precede both motor and non-motor symptoms.
In 1988, McGeer et al. [47] showed the presence of reactive microglia in the SNpc of human post-mortem brain tissues. High levels of interleukin-1β (IL1-β), IL-6, TNFα, interferon γ (IFNγ), cyclooxygenase type 2 (Cox-2), nitric oxide synthase (NOS), infiltrating peripheral immune cells, and ROS were found in brain tissues as well as in cerebrospinal fluids of PD patients. Moreover, a recent postmortem study on demented PD cases revealed a critical role of upregulation of neuroinflammatory markers, α-SYN pathology, and microglia [48][49]. Analysis of transcriptomic data from human postmortem brains showed the ubiquitous expression of TLR4. The expression of TLR4 was found higher in substantia nigra and putamen, in the circulating monocytes, and in the B cells of PD patients [50][51][52][53] Genetic studies show that TLR4 polymorphism is related to an increased risk of PD [54][55]. Also, increased TLR4 protein levels were detected in the peripheral immune cells, substantia nigra, and caudate/putamen of PD cases [53][56]. Studies in animal models of PD suggested the potential role of TLR4 in mediating biochemical changes as well as dopaminergic cell death and α-synuclein accumulation in the midbrain [57][58]. Moreover, TLR4 has been found to play a critical role as a mediator of the neurotoxicity induced by α-synuclein oligomers [59]. Additional studies in experimental models of PD and α-synucleinopathies showed the relevant involvement of TLR4 [60][61][62][63][64] TLR4 is expressed by two types of non-neuronal supportive cells: CNS residential macrophages or microglia and astrocytes. In the brain, TLR4-mediated signaling pathways have been implicated in the pathogenesis of PD [65]. Its stimulation causes the production of elevated levels of neurotoxic inflammatory cytokines that damage nigral dopaminergic neuron [66].
Drouin-Ouellet and co-workers [52] showed that the overexpression of α-SYN induced a critical modulation of TLR4 signaling in the blood and brain of both experimental models and PD patients. This triggers an immune/inflammatory response that culminates with neuronal death and negatively impacts motor symptoms. However, a study supported a role for TLR4 in mediating the clearance of α-SYN, suggesting an innate neuroprotective mechanism [67]. Therefore, both neuroprotective and detrimental roles of TLR4 in PD have been suggested: acute stimuli such as posttranslational modification of α-SYN can be a trigger for TLR4 microglial activation and protein clearance. Conversely, chronic inflammation, from inside or outside the nervous system, may promote the imbalanced activation of TLR4 signaling and escalation of inflammatory response which further contribute to the pathogenesis of PD [68][69]. A large number of molecules, as well as the several forms of α-SYN (monomer, oligomers, and fibrils, truncated and phosphorylated), released from neighboring neurons in the extracellular milieu, act as ligands for TLR4 and undergo phagocytosis, degradation and clearance. If compromised, this mechanism lead to further inflammatory signals responsible for the formation of α-SYN aggregated in the Lewy bodies of substantia nigra. In summary, it can be concluded that PD is the result of a-synuclein pathology and increased neuroinflammation in the nigro-striatal tract. In this context, TLR4 activation can play both neuroprotective and detrimental roles. Further studies are needed to investigate the clinical efficacy of targeting the TLR4 receptor for the management of PD.
References
- Vijay, K. “Toll-like receptors in immunity and inflammatory diseases: Past, present, and future.”Int. immunopharmacol. 2018;59:391-412. doi:10.1016/j.intimp.2018.03.002.
- Kawai T., Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637-50.
- Loiarro M., Ruggiero V., Sette C. Targeting TLR/IL-1R signalling in human diseases. Mediators Inflamm. 2010; 2010:674363.
- Barton G.M., Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524-5.
- Rajaiah R., Perkins D.J., Ireland D.D., Vogel S.N. CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc Natl Acad Sci U S A. 2015; 112:8391-6.
- Li Y., Deng S.L., Lian Z.X., Yu K. Roles of Toll-Like Receptors in Nitroxidative Stress in Mammals. Cells. 2019; 8:576. doi: 10.3390/cells8060576.
- Mhyre T.R., Boyd J.T., Hamill R.W., Maguire-Zeiss K.A. Parkinson’s disease. Subcell Biochem. 2012;65:389–455. doi: 10.1007/978-94-007-5416-4_16.
- Dorsey E.R., Bloem B.R. The Parkinson pandemic—A call to action.JAMA Neurol. 2018;75:9–10. doi:10.1001/jamaneurol.2017.3299.
- Moustafa A.A., Chakravarthy S., Phillips J.R., Gupta A., Keri S., Polner B. Motor symptoms in Parkinson’s disease: A unified framework. Neurosci. Biobehav. Rev. 2016;68:727–740. doi:10.1016/j.neubiorev.2016.07.010.
- Alexander G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004;6:259–280. doi: 10.31887/DCNS.2004.6.3/galexander.
- Moore D.J., West A.B., Dawson V.L., Dawson T.M. Molecular pathophysiology of Parkinson’s disease.Annu. Rev. Neurosci. 2005;28:57doi: 10.1146/annurev.neuro.28.061604.135718.
- Spillantini M.G., Schmidt M.L., Lee V.M., Trojanowski J.Q., Jakes R., Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi:10.1038/42166.
- Stefanis L. α-synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012;2:a009399. doi:10.1101/cshperspect.a009399.
- Kuan W.L., Stott K., He X., Wood T., Yang S., Kwok J., Hall K., Zhao Y., Tietz O., Aigbirhio F., et al. Systemic α-synuclein injection triggers selective neuronal pathology as seen in patients with Parkinson’s disease. Mol. Psychiatry.2021;26:556–567. doi:10.1038/s41380-019-0608-9.
- Ransohoff R.M., Kivisäkk P., Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003;3:569–581. doi:10.1038/nri1130.
- Chavan S.S., Pavlov V.A., Tracey K.J. Mechanisms and therapeutic relevance of neuro-immune communication.Immunity. 2017;46:927–942. doi:10.1016/j.immuni.2017.06.008.
- Hirsch E.C., Standaert D.G. Ten unsolved questions about neuroinflammation in Parkinson’s disease. Mov. Disord. 2021;36:16–24. doi:10.1002/mds.28075.
- Riederer P., Berg D., Casadei N., Cheng F., Classen J., Dresel C., Jost W., Krüger R., Müller T., Reichmann H., et al. α-Synuclein in Parkinson’s disease: Causal or bystander? J. Neural Transm. 2019;126:815–840. doi:10.1007/s00702-019-02025-9.
- Jellinger K.A. Is Braak staging valid for all types of Parkinson’s disease? J. Neural Transm. 2019;126:423–431. doi:10.1007/s00702-018-1898-9.
- Sian-Hulsmann J., Riederer P. The nigral coup in Parkinson’s disease by α-synuclein and its associated rebels. Cells. 2021;10:598. doi:10.3390/cells10030598.
- Villegas-Llerena C., Phillips A., Garcia-Reitboeck P., Hardy J., Pocock J.M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 2016;36:74–81. doi:10.1016/j.conb.2015.10.004.
- Philips T., Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:235–263. doi: 10.1016/S1474-4422(11)70015-1.
- Spargo E., Everall I.P., Lantos P.L. Neuronal loss in the hippocampus in Huntington’s disease: A comparison with HIV infection. J. Neurol. Neurosurg. Psychiatry. 1993;56:487–491. doi: 10.1136/jnnp.56.5.487.
- Giralt A., Saavedra A., Alberch J., Pérez-Navarro E. Cognitive dysfunction in Huntington’s disease: Humans, mouse models and molecular mechanisms. J. Huntingt. Dis. 2012;1:155–173. doi: 10.3233/JHD-120023.
- Alam A., Thelin E.P., Tajsic T., Khan D.Z., Khellaf A., Patani R., Helmy A. Cellular infiltration in traumatic brain injury. J. Neuroinflamm. 2020;17:328. doi: 10.1186/s12974-020-02005-x.
- Norden D.M., Trojanowski P.J., Villanueva E., Navarro E., Godbout J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia. 2016;64:300–316. doi: 10.1002/glia.22930.
- Schetters S.T.T., Gomez-Nicola D., Garcia-Vallejo J.J., Van Kooyk Y. Neuroinflammation: Microglia and T cells get ready to tango. Front. Immunol. 2018;8:1905. doi: 10.3389/fimmu.2017.01905.
- Sulzer D., Alcalay R.N., Garretti F., Cote L., Kanter E., Agin-Liebes J., Liong C., McMurtrey C., Hildebrand W.H., Mao X., et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature. 2017;546:656–661. doi: 10.1038/nature22815.
- Colonna M., Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017;35:441–468. doi: 10.1146/annurev-immunol-051116-052358.
- Sevenich L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front. Immunol. 2018;9:697. doi: 10.3389/fimmu.2018.00697.
- Rueda-Carrasco J., Hong S. The jekyll and hyde of TREM2. Trends Neurosci. 2020;43:739–740. doi: 10.1016/j.tins.2020.08.002.
- Augusto-Oliveira M., Arrifano G.P., Lopes-Araújo A., Santos-Sacramento L., Takeda P.Y., Anthony D.C., Malva J.O., Crespo-Lopez M.E. What do microglia really do in healthy adult brain? Cells. 2019;8:1293. doi: 10.3390/cells8101293.
- Liu H., Leak R.K., Hu X. Neurotransmitter receptors on microglia. Stroke Vasc. Neurol. 2016;1:52–58. doi: 10.1136/svn-2016-000012.
- Nayak D., Roth T.L., McGavern D.B. Microglia development and function. Ann. Rev. Immunol. 2014;32:367–402. doi: 10.1146/annurev-immunol-032713-120240.
- Arcuri C., Mecca C., Bianchi R., Giambanco I., Donato R. The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front. Mol. Neurosci. 2017;10:191. doi: 10.3389/fnmol.2017.00191.
- Lecours C., Bordeleau M., Cantin L., Parent M., Di Paolo T., Tremblay M.E. Microglial implication in Parkinson’s disease: Loss of beneficial physiological roles or gain of inflammatory functions? Front. Cell Neurosci. 2018;12:282–289. doi: 10.3389/fncel.2018.00282.
- Tan J.S.Y., Chao Y.X., Rötzschke O., Tan E.K. New insights into immune-mediated mechanisms in Parkinson’s disease. Int. J. Mol. Sci. 2020;21:9302. doi: 10.3390/ijms21239302.
- Bartels A.L., Leenders K.L. Cyclooxygenase and neuroinflammation in Parkinsons disease neurodegeneration. Curr. Neuropharmacol. 2010;8:62. doi: 10.2174/157015910790909485.
- Gerhard A., Pavese N., Hotton G., Turkheimer F., Es M., Hammers A., Eggert K., Oertel W., Banati R.B., Brooks D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiology. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002.
- Lee P.C., Artaud F., Cormier-Dequaire F., Rascol O., Durif F., Derkinderen P. Examining the reserve hypothesis in Parkinson’s disease: A longitudinal study. Mov. Disord. 2019;34:1663–1671. doi: 10.1002/mds.27854.
- Pajares M.I., Rojo A., Manda G., Boscá L., Cuadrado A. Inflammation in Parkinson’s disease: Mechanisms and therapeutic implications. Cells. 2020;9:1687. doi: 10.3390/cells9071687.
- Marogianni C., Sokratous M., Dardiotis E., Hadjigeorgiou G.M., Bogdanos D., Xiromerisiou G. Neurodegeneration and Inflammation—An Interesting interplay in Parkinson’s disease. Int. J. Mol. Sci. 2020;21:8421. doi: 10.3390/ijms21228421.
- Chagas L., Sandre P.C., Ribeiro E., Ribeiro N., Marcondes H., Oliveira Silva P. environmental signals on microglial function during brain development, neuroplasticity, and disease. Int. J. Mol. Sci. 2020;21:2111. doi: 10.3390/ijms21062111.
- Kettenmann H., Hanisch U.K., Noda M., Verkhratsky A. Physiology of microglia. Physiol. Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010.
- Hammond T.R., Robinton D., Stevens B. Microglia and the brain: Complementary partners in development and disease. Ann. Rev. Cell Dev. Biol. 2018;34:523–544. doi: 10.1146/annurev-cellbio-100616-060509.
- Davalos D., Grutzendler J., Yang G., Kim J.V., Zuo Y., Jung S., Littman D.R., Dustin M.L., Gan W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005;8:752–758. doi: 10.1038/nn1472.
- McGeer P.L., Itagaki S., Boyes B.E., McGeer E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/WNL.38.8.1285.
- Mogi M., Harada M., Kondo T., Riederer P., Inagaki H., Minami M., Nagatsu T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994;180:147–150. doi:10.1016/0304-3940(94)90508-8.
- Kouli A., Camacho M., Allinson K., Williams-Gray C.H. Neuroinflammation and protein pathology in Parkinson’s disease dementia.Acta Neuropathol. Commun. 2020;8:211. doi:10.1186/s40478-020-01083-5.
- Gelders G., Baekelandt V., Van der Perren A. Linking Neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018:4784268. doi: 10.1155/2018/4784268.
- Refolo V., Stefanova N. Neuroinflammation and glial phenotypic changes in alpha-synucleinopathies. Front. Cell Neurosci. 2019;13:263. doi: 10.3389/fncel.2019.00263.
- Drouin-Ouellet J., St-Amour I., Saint-Pierre M., Lamontagne-Proulx J., Kriz J., Barker R.A., Cicchetti F. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int. J. Neuropsychopharmacol. 2014;18:pyu103. doi: 10.1093/ijnp/pyu103.
- Doorn K.J., Moors T., Drukarch B., van de Berg W.D.J., Lucassen P.J., van Dam A.M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014;2:90. doi: 10.1186/s40478-014-0090-1.
- Bsibsi M., Ravid R., Gveric D., van Noort J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013.
- Zhao J., Han X., Xue L., Zhu K., Liu H., Xie A. Association of TLR4 gene polymorphisms with sporadic Parkinson’s disease in a Han Chinese population. Neurol. Sci. 2015;36:1659–1665. doi: 10.1007/s10072-015-2227-9.
- Hughes C.D., Choi M.L., Ryten M., Hopkins L., Drews A., Botía J.A., Iljina M., Rodrigues M., Gagliano S.A., Gandhi S., et al. Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol. 2019;137:103–120. doi: 10.1007/s00401-018-1907-y.
- Mariucci G., Pagiotti R., Galli F., Romani L., Conte C. The potential role of toll-like receptor 4 in mediating dopaminergic cell loss and alpha-synuclein expression in the acute MPTP mouse model of Parkinson’s disease. J. Mol. Neurosci. 2018;64:611–618. doi: 10.1007/s12031-018-1057-7.
- Conte C., Roscini L., Sardella R., Mariucci G., Scorzoni S., Beccari T., Corte L. Toll like receptor 4 affects the cerebral biochemical changes induced by MPTP treatment. Neurochem. Res. 2017;42:493–500. doi: 10.1007/s11064-016-2095-6.
- Rannikko E.H., Weber S.S., Kahle P.J. Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015;16:57. doi:10.1186/s12868-015-0192-0.
- Rueda-Carrasco J., Hong S. The jekyll and hyde of TREM2. Trends Neurosci. 2020;43:739–740. doi: 10.1016/j.tins.2020.08.002.
- Augusto-Oliveira M., Arrifano G.P., Lopes-Araújo A., Santos-Sacramento L., Takeda P.Y., Anthony D.C., Malva J.O., Crespo-Lopez M.E. What do microglia really do in healthy adult brain?Cells.2019;8:1293. doi: 10.3390/cells8101293.
- Shin W.H., Jeon M.T., Leem E., Won S.Y., Jeong K.H., Park S.J., McLean C., Lee S.J., Jin B.K., Jung U.J., et al. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: A potential pathogenic mechanism in Parkinson’s disease. Sci. Rep. 2015;5:14764. doi: 10.1038/srep14764.
- Lehnardt S., Massillon L., Follett P., Jensen F.E., Ratan R., Rosenberg P.A. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100.
- Panaro M.A., Lofrumento D.D., Saponaro C., De Nuccio F., Cianciulli A., Mitolo V., Nicolardi G. Expression of TLR4 and CD14 in the central nervous system (CNS) in a MPTP mouse model of Parkinson’s-like disease. Immunopharmacol. Immunotoxicol. 2008;30:729–740. doi: 10.1080/08923970802278557.
- Fellner L., Irschick R., Schanda K., Reindl M., Klimaschewski L., Poewe W., Wenning G.K., Stefanova N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia.2013;61:349–360. doi:10.1002/glia.22437.
- Olson J.K., Miller S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs.J. Immunol. 2004;173:3916–3924. doi:10.4049/jimmunol.173.6.3916.
- Stefanova N., Fellner L., Reindl M., Masliah E., Poewe W., Wenning G.K. Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol.2011;179:954–963. doi:10.1016/j.ajpath.2011.04.013.
- Leitner G.R., Wenzel T.J., Marshall N., Gates E.J., Klegeris A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther.Targ. 2019;23:865–882. doi:10.1080/14728222.2019.1676416.
- Campolo C., Paterniti I., Siracusa R., Filippone A., Esposito E., Cuzzocrea S. TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav. Immun. 2019;76:236–247. doi:10.1016/j.bbi.2018.12.003.


