 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Xin Wu | + 1409 word(s) | 1409 | 2021-09-14 12:32:56 | | | |
| 2 | Beatrix Zheng | + 515 word(s) | 1924 | 2021-09-24 05:44:17 | | |
Video Upload Options
Calmodulin (CaM) is a small protein that acts as a ubiquitous signal transducer and regulates neuronal plasticity, muscle contraction, and immune response. It interacts with ion channels and plays regulatory roles in cellular electrophysiology. CaM modulates the voltage-gated sodium channel gating process, alters sodium current density, and regulates sodium channel protein trafficking and expression.
1. Introduction
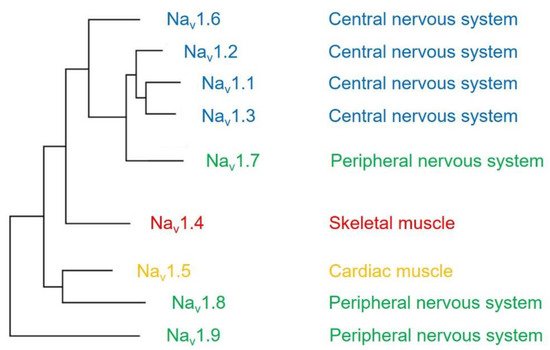
The voltage-gated sodium channel (Na v ) plays a vital role in the generation and propagation of action potential in excitable cells such as neurons and cardiac myocytes [1]. The family of voltage-gated sodium channels has nine members named Na v 1.1 through Na v 1.9 [2]. Among them, Na v 1.1, Na v 1.2, Na v 1.3, and Na v 1.6 are predominantly expressed in neurons of the central nervous system [3][4]. Three isoforms (Na v 1.7, Na v 1.8, and Na v 1.9) are widely expressed in neurons of the peripheral nervous system, such as dorsal root ganglia (DRG) neurons [5][6][7]. The isoform Na v 1.4 is responsible for upstroke of the action potential in skeletal muscle [8], and Na v 1.5 is a cardiac-specific isoform known as cardiac sodium channel ( Figure 1 ) [9].
An essential property of voltage-gated sodium channels is “inactivation”, which prevents the reopening of the channel until complete recovery [10]. The inactivation regulates the frequency of action potential firing initiated by sodium channels in excitable cells. It reduces the breakdown of ionic gradients and cell death.
All voltage-gated sodium channels contain a calmodulin (CaM)-binding IQ domain necessary for the channel inactivation [11]. CaM is a small protein expressed in all eukaryotic cells [12]. It acts as a ubiquitous signal transducer and regulates essential processes such as neuronal plasticity, muscle contraction, and immune response [13]. CaM has two globular domains (i.e., N-terminal lobe and C-terminal lobe) connected by a linker. Each lobe contains two EF-hand motifs binding to Ca 2+ [14]. In the Ca 2+ -free state, the EF-hands are collapsed in a compact configuration. When Ca 2+ is bound to CaM, a conformational change occurs in the protein and rearranges the structural information of CaM.
The CaM-binding IQ domain is located within the C-terminal domain of the voltage-gated sodium channel. It contains around 25 residues with two highly conserved amino acids, isoleucine (I) and glutamine (Q), in the middle of the motif. The residues in the IQ domain form a seven-turn α-helix binding to CaM in a Ca 2+ -independent manner. In addition to the IQ domain, a globular domain in the C-terminus of voltage-gated sodium channel consists of EF hand-like (EFL) motifs that interact with CaM in the absence of Ca 2+ [15][16], whereas an intracellular loop connecting domains III and IV (III-IV linker) in some isoforms of the sodium channel is shown to interact with CaM in the presence of Ca 2+ [17].
2. CaM Regulation of Voltage-Gated Sodium Channel
CaM binds to the voltage-gated sodium channel and modulates the sodium channel gating process [15], alters sodium current density [18], and regulates sodium channel protein trafficking and expression [19].
One of the CaM modulations of voltage-gated sodium channel gating is inactivation ( Figure 2 ). Voltage-gated sodium channel has two different types of inactivation, known as fast inactivation and slow inactivation. Fast inactivation occurs by occlusion of the intracellular pore by an “inactivation gate” formed by the cytoplasmic III-IV linker [10][20]. Slow inactivation is related to conformational rearrangements of the selectivity filter of the channel [21][22]. CaM mediates the fast inactivation by an interaction between III-IV linker and C-terminal domain of the voltage-gated sodium channel [23]. It enhances the slow inactivation by inducing a hyperpolarized shift of voltage dependence of inactivation and reducing the channel’s availability. CaM interaction with the sodium channel affects channel inactivation kinetics. It strengthens the development of inactivation and slows the recovery from inactivated state of the sodium channel.
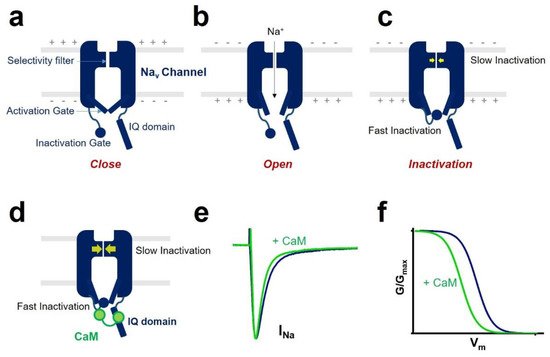
In some isoforms of the voltage-gated sodium channels, CaM influences the activation process. It was reported that co-expression of CaM and Na v 1.1 produced a hyperpolarized shift of the voltage dependence of activation of the Na v 1.1 sodium channel [24].
Moreover, CaM can modulate sodium channel current density [18]. Studies reported that CaM regulation of sodium current density is Ca 2+ -dependent [24][25]. CaM does not affect sodium current amplitude in the absence of Ca 2+ . In contrast, it increases the current density in the presence of a high concentration of Ca 2+ .
3. Modulators Involved in CaM Regulation of Sodium Channel
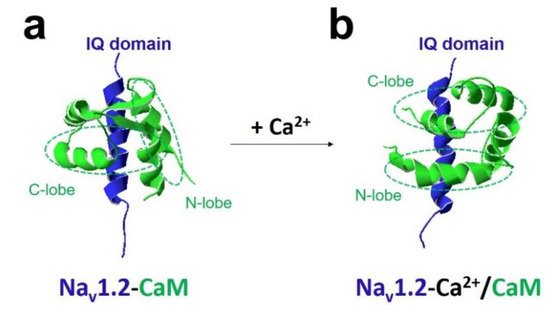
The most crucial regulator is Ca 2+ [11][26]. In the presence of Ca 2+ , Ca 2+ /CaM (Ca 2+ -binding form of CaM) has a different regulation of sodium channel function compared with apo-CaM (Ca 2+ free CaM), and Ca 2+ binding to CaM induces rearrangements between the voltage-gated sodium channel and CaM lobes. In the absence of Ca 2+ , the N-lobe of CaM does not target the IQ domain ( Figure 3 a and Figure 4 a,b). In the presence of Ca 2+ , the structure of the N-lobe is rearranged by Ca 2+ . It generates an allosterically conformational change binding to the distal IQ domain of the channel ( Figure 3 b and Figure 4 c). However, the C-lobe of CaM can bind to the voltage-gated sodium channel IQ domain in a Ca 2+ -independent manner.
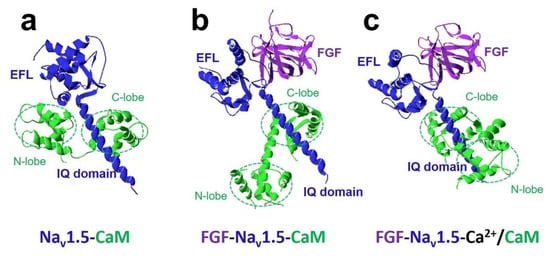
The FHFs were shown to form a complex with CaM and modulate CaM regulation of Na v 1.5 function [27]. A recent study discovered fibroblast growth factor homologous factor FGF13 (fibroblast growth factor 13, also known as FHF2) tuned arrhythmogenic late sodium currents in CaM binding-deficient channels in cardiac myocytes [27]. The crystal structures in Figure 4 showed that FGF and Ca 2+ mediated the interaction between CaM and Na v 1.5 IQ domain. It indicated that the N-lobe of CaM interacted with the EFL of the channel in the absence of FGF. However, when FGF was bound to the channel, the N-lobe of CaM was rearranged. It did not make contact with the EFL and was free to interact with other parts of the channel complex. The N-lobe of CaM likely interacted with the III-IV linker of the sodium channel and modulated the channel’s inactivation.
In addition, CaMKII has been reported to influence CaM interaction with the sodium channel. One study found CaMKII-mediated phosphorylation of the Na v 1.1 sodium channel IQ domain increased CaM binding affinity to the channel [28]. Three phosphorylation sites (T1909, S1918, and T1934) were identified in the Na v 1.1 IQ domain. Other studies demonstrated that CaMKII phosphorylated Na v 1.5 at residue S571 to decrease channel availability. The CaMKII phosphorylation site S571 is located at the intracellular DI-DII loop of the Na v 1.5 sodium channel, and mutation at position S571 abolished the hyperpolarized shift of the voltage dependence of inactivation produced by CaMKII phosphorylation. The effects of CaMKII-mediated phosphorylation of IQ domain on sodium currents have not been characterized. CaMKII-dependent IQ domain phosphorylation might confer normal sodium currents important for the function of cells where the Na v channels are expressed. The CaMKII-mediated effect seems to be isoform-specific. CaMKII phosphorylation enhances CaM affinity for sodium channel Na v 1.1 but not for Na v 1.2 [28][29] .
With these partners, CaM regulates voltage-gated sodium channel function by altering channel gating, sodium current density, and expression of the sodium channel protein. Studies have shown that CaM has isoform-specific modulation of voltage-gated sodium channel function [30][31][32][33].
4. Interaction between CaM and Sodium Channel Isoforms
Na v 1.4 is the predominant subtype of sodium channel initiating skeletal muscle action potential [8]. Na v 1.4 mutations associated with periodic paralysis disorders affect skeletal muscle excitability [34].
The sodium channel Na v 1.5 IQ domain mutations associated with arrhythmia reduce CaM binding affinity. In HEK293 cells, overexpression of CaM attenuates late sodium currents caused by Na v 1.5 IQ domain mutations [35]. CaM binding to Na v 1.5 channel IQ domain also modifies late sodium currents in cardiac myocytes [27]. Study in transgenic mouse models showed FHFs tuned cardiac late sodium currents in ventricular myocytes. FHFs diminished late sodium current of the Na v 1.5 channel IQ/AA mutation (substitution of two conserved residues isoleucine (I) and glutamine (Q) with double alanines (AA) in the IQ domain), IQ/AA mutation has been reported to disrupt CaM binding to the IQ domain of voltage-gated sodium channels.
Studies in guinea-pig ventricular myocytes revealed CaM enhances the cardiac sodium current density [36]; this is different from the result that CaM does not influence on sodium current amplitude when co-expressing with Na v 1.5 channel in HEK293 cells [35]. It indicates that some environmental factors in cardiac tissues are involved in CaM regulation of Na v function.
To date, there is no study investigating the interaction between CaM and Na v 1.9 channel. The Na v 1.9 IQ domain shares high sequence homology with other sodium channels ( Figure 5 ). It remains to be determined if CaM regulation of Na v 1.9 channel plays a role in channel function in sensory neurons.

References
- Xu, L.; Ding, X.; Wang, T.; Mou, S.; Sun, H.; Hou, T. Voltage-gated sodium channels: Structures, functions, and molecular modeling. Drug Discov. Today 2019, 24, 1389–1397.
- Wood, J.N.; Iseppon, F. Sodium channels. Brain Neurosci. Adv. 2018, 2, 2398212818810684.
- Encinas, A.C.; Watkins, J.C.; Longoria, I.A.; Johnson, J.P.; Hammer, M.F. Variable patterns of mutation density among NaV1.1, NaV1.2 and NaV1.6 point to channel-specific functional differences associated with childhood epilepsy. PLoS ONE 2020, 15, e0238121.
- Wang, Z.; Kuang, P.; Lin, Y.; Liu, W.; Lao, W.; Ji, Y.; Zhu, H. Re-expression of voltage-gated sodium channel subtype Nav1.3 in the substantia nigra after dopamine depletion. Neurosci. Lett. 2018, 687, 146–152.
- Wang, J.T.; Zheng, Y.M.; Chen, Y.T.; Gu, M.; Gao, Z.B.; Nan, F.J. Discovery of aryl sulfonamide-selective Nav1.7 inhibitors with a highly hydrophobic ethanoanthracene core. Acta Pharmacol. Sin. 2020, 41, 293–302.
- Xiao, Y.; Barbosa, C.; Pei, Z.; Xie, W.; Strong, J.A.; Zhang, J.M.; Cummins, T.R. Increased Resurgent Sodium Currents in Nav1.8 Contribute to Nociceptive Sensory Neuron Hyperexcitability Associated with Peripheral Neuropathies. J. Neurosci. 2019, 39, 1539–1550.
- Bai, Q.; Shao, J.; Cao, J.; Ren, X.; Cai, W.; Su, S.; George, S.; Tan, Z.; Zang, W.; Dong, T. Protein kinase C-alpha upregulates sodium channel Nav1.9 in nociceptive dorsal root ganglion neurons in an inflammatory arthritis pain model of rat. J. Cell Biochem. 2020, 121, 768–778.
- Mannikko, R.; Wong, L.; Tester, D.J.; Thor, M.G.; Sud, R.; Kullmann, D.M.; Sweeney, M.G.; Leu, C.; Sisodiya, S.M.; FitzPatrick, D.R.; et al. Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: A case-control study. Lancet 2018, 391, 1483–1492.
- Turan, N.N.; Moshal, K.S.; Roder, K.; Baggett, B.C.; Kabakov, A.Y.; Dhakal, S.; Teramoto, R.; Chiang, D.Y.; Zhong, M.; Xie, A.; et al. The endosomal trafficking regulator LITAF controls the cardiac Nav1.5 channel via the ubiquitin ligase NEDD4-2. J. Biol. Chem. 2020, 295, 18148–18159.
- Nakajima, T.; Kaneko, Y.; Dharmawan, T.; Kurabayashi, M. Role of the voltage sensor module in Nav domain IV on fast inactivation in sodium channelopathies: The implication of closed-state inactivation. Channels (Austin) 2019, 13, 331–343.
- Gabelli, S.B.; Yoder, J.B.; Tomaselli, G.F.; Amzel, L.M. Calmodulin and Ca(2+) control of voltage gated Na(+) channels. Channels (Austin) 2016, 10, 45–54.
- O’Day, D.H.; Taylor, R.J.; Myre, M.A. Calmodulin and Calmodulin Binding Proteins in Dictyostelium: A Primer. Int J. Mol. Sci. 2020, 21, 1210.
- Zoidl, G.R.; Spray, D.C. The Roles of Calmodulin and CaMKII in Cx36 Plasticity. Int J. Mol. Sci. 2021, 22, 4473.
- Villalobo, A.; Ishida, H.; Vogel, H.J.; Berchtold, M.W. Calmodulin as a protein linker and a regulator of adaptor/scaffold proteins. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 507–521.
- Kang, P.W.; Chakouri, N.; Diaz, J.; Tomaselli, G.F.; Yue, D.T.; Ben-Johny, M. Elementary mechanisms of calmodulin regulation of NaV1.5 producing divergent arrhythmogenic phenotypes. Proc. Natl. Acad. Sci. USA 2021, 118.
- Gabelli, S.B.; Boto, A.; Kuhns, V.H.; Bianchet, M.A.; Farinelli, F.; Aripirala, S.; Yoder, J.; Jakoncic, J.; Tomaselli, G.F.; Amzel, L.M. Regulation of the NaV1.5 cytoplasmic domain by calmodulin. Nat. Commun. 2014, 5, 5126.
- Sarhan, M.F.; Tung, C.C.; Van Petegem, F.; Ahern, C.A. Crystallographic basis for calcium regulation of sodium channels. Proc. Natl. Acad. Sci. USA 2012, 109, 3558–3563.
- Choi, J.S.; Hudmon, A.; Waxman, S.G.; Dib-Hajj, S.D. Calmodulin regulates current density and frequency-dependent inhibition of sodium channel Nav1.8 in DRG neurons. J. Neurophysiol. 2006, 96, 97–108.
- Biswas, S.; Deschenes, I.; Disilvestre, D.; Tian, Y.; Halperin, V.L.; Tomaselli, G.F. Calmodulin regulation of Nav1.4 current: Role of binding to the carboxyl terminus. J. Gen. Physiol. 2008, 131, 197–209.
- Li, Z.; Jin, X.; Wu, T.; Zhao, X.; Wang, W.; Lei, J.; Pan, X.; Yan, N. Structure of human Nav1.5 reveals the fast inactivation-related segments as a mutational hotspot for the long QT syndrome. Proc. Natl. Acad. Sci. USA 2021, 118.
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu Rev. Pharmacol Toxicol. 2020, 60, 133–154.
- Chatterjee, S.; Vyas, R.; Chalamalasetti, S.V.; Sahu, I.D.; Clatot, J.; Wan, X.; Lorigan, G.A.; Deschenes, I.; Chakrapani, S. The voltage-gated sodium channel pore exhibits conformational flexibility during slow inactivation. J. Gen. Physiol. 2018, 150, 1333–1347.
- Gade, A.R.; Marx, S.O.; Pitt, G.S. An interaction between the III-IV linker and CTD in NaV1.5 confers regulation of inactivation by CaM and FHF. J. Gen. Physiol. 2020, 152.
- Gaudioso, C.; Carlier, E.; Youssouf, F.; Clare, J.J.; Debanne, D.; Alcaraz, G. Calmodulin and calcium differentially regulate the neuronal Nav1.1 voltage-dependent sodium channel. Biochem. Biophys Res. Commun. 2011, 411, 329–334.
- Rusconi, R.; Scalmani, P.; Cassulini, R.R.; Giunti, G.; Gambardella, A.; Franceschetti, S.; Annesi, G.; Wanke, E.; Mantegazza, M. Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant. J. Neurosci. 2007, 27, 11037–11046.
- Johnson, C.N. Calcium modulation of cardiac sodium channels. J. Physiol. 2020, 598, 2835–2846.
- Abrams, J.; Roybal, D.; Chakouri, N.; Katchman, A.N.; Weinberg, R.; Yang, L.; Chen, B.X.; Zakharov, S.I.; Hennessey, J.A.; Avula, U.M.R.; et al. Fibroblast growth factor homologous factors tune arrhythmogenic late NaV1.5 current in calmodulin binding-deficient channels. JCI Insight 2020, 5.
- Li, J.; Yu, Z.; Xu, J.; Feng, R.; Gao, Q.; Boczek, T.; Liu, J.; Li, Z.; Wang, Q.; Lei, M.; et al. The Effect of Ca(2+), Lobe-Specificity, and CaMKII on CaM Binding to NaV1.1. Int J. Mol. Sci. 2018, 19, 2495.
- Jia, W.; Liu, J.; Yu, Z.; Zhang, X.; Xu, X.; Wang, Y.; Gao, Q.; Feng, R.; Wan, Y.; Xu, J.; et al. Properties of Calmodulin Binding to NaV1.2 IQ Motif and Its Autism-Associated Mutation R1902C. Neurochem. Res. 2021, 46, 523–534.
- Deschenes, I.; Neyroud, N.; DiSilvestre, D.; Marban, E.; Yue, D.T.; Tomaselli, G.F. Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin. Circ. Res. 2002, 90, E49–E57.
- Herzog, R.I.; Liu, C.; Waxman, S.G.; Cummins, T.R. Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J. Neurosci. 2003, 23, 8261–8270.
- Ben-Johny, M.; Yang, P.S.; Niu, J.; Yang, W.; Joshi-Mukherjee, R.; Yue, D.T. Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels. Cell. 2014, 157, 1657–1670.
- Yoder, J.B.; Ben-Johny, M.; Farinelli, F.; Srinivasan, L.; Shoemaker, S.R.; Tomaselli, G.F.; Gabelli, S.B.; Amzel, L.M. Ca(2+)-dependent regulation of sodium channels NaV1.4 and NaV1.5 is controlled by the post-IQ motif. Nat. Commun. 2019, 10, 1514.
- Horie, R.; Kubota, T.; Koh, J.; Tanaka, R.; Nakamura, Y.; Sasaki, R.; Ito, H.; Takahashi, M.P. EF hand-like motif mutations of Nav1.4 C-terminus cause myotonic syndrome by impairing fast inactivation. Muscle Nerve 2020, 61, 808–814.
- Yan, H.; Wang, C.; Marx, S.O.; Pitt, G.S. Calmodulin limits pathogenic Na+ channel persistent current. J. Gen. Physiol. 2017, 149, 277–293.
- Aiba, T.; Hesketh, G.G.; Liu, T.; Carlisle, R.; Villa-Abrille, M.C.; O’Rourke, B.; Akar, F.G.; Tomaselli, G.F. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc. Res. 2010, 85, 454–463.


