 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cristiano Simone | + 3864 word(s) | 3864 | 2021-09-08 07:45:23 | | | |
| 2 | Vicky Zhou | Meta information modification | 3864 | 2021-09-14 05:10:35 | | |
Video Upload Options
The SMYD3 methyltransferase has been found overexpressed in several types of cancers of the gastrointestinal (GI) tract. While high levels of SMYD3 have been positively correlated with cancer progression in cellular and advanced mice models, suggesting it as a potential risk and prognosis factor, its activity seems dispensable for autonomous in vitro cancer cell proliferation. We first describe the oncogenic activity of SMYD3 as a transcriptional activator of genes involved in tumorigenesis, cancer development and transformation and as a co-regulator of key cancer-related pathways. Then, we dissect its role in orchestrating cell cycle regulation and DNA damage response (DDR) to genotoxic stress by promoting homologous recombination (HR) repair, thereby sustaining cancer cell genomic stability and tumor progression. Based on this evidence and on the involvement of PARP1 in other DDR mechanisms, we also outline a synthetic lethality approach consisting of the combined use of SMYD3 and PARP inhibitors, which recently showed promising therapeutic potential in HR-proficient GI tumors expressing high levels of SMYD3. Overall, these findings identify SMYD3 as a promising target for drug discovery.
1. Introduction
SMYD3 is a member of the SMYD (SET and MYND Domain) lysine methyltransferase family, which includes five members (SMYD1-5) [1]. Their methyltransferase activity requires the combination of the SET domain with adjacent cysteine-rich regions, one located N-terminally (pre-SET or N-SET) and the other posterior to the SET domain (post-SET). Pre- and post-SET domains seem to play a crucial role in the substrate recognition and enzymatic activity of SMYD family members [1][2]. The MYND domain is the structural discriminant between SMYDs and other SET domain-containing proteins and is found in several transcriptional regulators, in which it facilitates the interactions with partner proteins through PXLXP motifs [3]. Structural analyses showed that the N-terminal region of human SMYD3 includes the SET, MYND, and post-SET domains, while the C-terminal region contains a tetratricopeptide repeat (TPR)-like domain that modulates SMYD3 interaction with the consensus motif MEEVD of HSP90 and other proteins, and its nuclear localization [1][4][5].
SMYD3 was first characterized as a histone H3 lysine 4 (H3K4) methyltransferase, with studies carried out in SMYD3-knocked down cell cultures confirming that its genetic ablation was often associated with reduced H3K4 methylation [6][7][8][9]. On the other hand, Van Aller and colleagues demonstrated that the preferred target of SMYD3-mediated methylation in vitro is H4K5 [10], which is consistent with results from our group [9].
Subsequent studies revealed that SMYD3 is an important epigenetic regulator that acts in both the nuclear and the cytoplasmic compartments and can interact with and methylate both histone and non-histone proteins. At the nuclear level, SMYD3 was initially shown to be recruited at CCTCCC DNA sequences [6]. Subsequently, genome-wide approaches revealed that it binds to DNA at target regions of transcription factors involved in cell proliferation [11]. Furthermore, SMYD3 is a crucial member of the transcriptional complex formed by RNA polymerase II and the RNA helicase HELZ [6] and serves as a coactivator of transcription processes [12][13][14]. In the cytoplasm, SMYD3 has been found to affect key factors involved in oncogenic pathways by interacting with and methylating non-histone proteins, which suggests a role as a modulator of signaling cascades promoting tumor progression [15][16][17][18].
SMYD3 activity does not appear to be required for normal development, as demonstrated by recent studies in SMYD3 knockout (KO) mice [11][15]. These results were confirmed in male and female SMYD3 homozygous conditional KO mice, which did not show significant abnormalities after whole phenotyping [19]. It has been reported that SMYD3 overexpression in normal cells is sufficient to accelerate cell growth and trigger the activation of genes involved in the transformation and migration of cancer cells [7][8]. Consistently, a close correlation has been observed between SMYD3 activity and the development of several malignancies. Indeed, SMYD3 has been found overexpressed in several types of cancers, including colorectal (CRC), breast (BC), gastric (GC), pancreatic (PC), ovarian (OvCa), prostate, and lung cancer, and hepatocellular carcinoma (HCC) [1], with high SMYD3 levels being associated with reduced overall survival and worse prognosis [11][20][21][22][23][24][25].
Recent studies provided evidence that SMYD3 may be an important biomarker for the diagnosis of several types of cancers and a potential target for drug discovery [1]. As a result, several SMYD3 chemical compounds able to inhibit its enzymatic activity were recently developed [26]. Through a virtual screening, in 2015 we identified the first substrate competitive SMYD3 inhibitor (SMYD3i) named BCI-121, which showed antigrowth properties and confirm the potential of targeting this protein [9]. Then, three more potent reversible SMYD3is (EPZ031686, EPZ030456, and, later, EPZ028862) were developed by the biopharmaceutical company Epizyme, which had a nanomolar potency and therefore a potential for in vivo assays [27][28]. Another SMYD3i was also described, named GSK2807, which acts as a competitive ligand at the cofactor binding site [29]. Moreover, the existing drug Diperodon has been reported as a new allosteric ligand interacting with SMYD3, representing a good starting point for design of tool compounds interacting with a druggable allosteric site, as modulators of noncatalytic SMYD3 functions [30]. In addition to these, current studies are revealing new selective SMYD3is (i.e. BAY-6530, covalent inhibitors 1-4) [26][31], thereby contributing to the ongoing identification of new effective SMYD3is as anticancer drugs.
2. SMYD3 Oncogenic Functions
Growing evidence supports a key role for SMYD3 in tumorigenesis in several cancer types. SMYD3 has been found to exert its oncogenic effects through transcriptional activation of a set of downstream target genes involved in cell death and proliferation (e.g., hTERT, WNT10B) [32][33], epithelial-mesenchymal transition (EMT) (e.g., SLUG, MMP2, MET) [11][20][21][34][35][36], cell cycle regulation (e.g., CCNA1, CCNA2, CCND1, CCNE1, PCNA, CDK2) [11][14][21][37], stem cell maintenance (e.g., ASCL2) [38], as well as oncogenes (e.g., MYC, JAK1/2, CTNNB1) [11]. Several studies investigated the mechanisms by which SMYD3 promotes the transcription of target genes, showing that it can directly occupy their promoter regions, interact with the transcriptional machinery, form complexes with RNA polymerase II [6][11] and other coactivators, such as PC4 [13], and associate with active chromatin by interacting with H3K4me3 tails [11]. Furthermore, it has been reported that SMYD3 dimethylates and colocalizes with the histone variant H2A.Z.1 at the promoter of the CCNA1 gene, inducing its expression and G1/S progression [14].
Besides regulating gene expression, SMYD3 has been shown to play a significant role in human cancer by modulating various key cancer-associated factors and therefore their related oncogenic pathways. Intriguingly, several studies revealed that SMYD3 exerts its oncogenic role primarily by interacting with and methylating non-histone proteins, through which it transactivates specific pathways involved in the survival and expansion of cancer cells [5][12][15][16][17]. In lung cancers and PCs, SMYD3 has a pivotal role in the regulation of oncogenic RAS signaling through the methylation of MAP3K2 kinase on lysine 206, which induces MAP3K2 release from the negative regulator PP2A phosphatase complex and therefore promotes ERK1/2 phosphoactivation [15]. Consistent with these findings, SMYD3 deletion or pharmacological inhibition resulted in lower ERK1/2 phosphorylation and thus reduced MEK-ERK signaling and tumor development in response to oncogenic RAS in CRCs and PCs [9][15]. In addition, SMYD3 can methylate lysine 14 on the AKT1 kinase, which promotes its phosphoactivation and plasma membrane accumulation, suggesting that SMYD3 methyltransferase activity may trigger the constitutive activation of AKT1 in cancer cells [16]. SMYD3 was also reported to interact with the estrogen receptor (ER) and potentiate ER-driven transcription, thereby promoting ER-mediated tumorigenicity [12]. It has been further shown that SMYD3 interaction with p53, which promotes p53 translocation into the cytoplasm and subsequent degradation, and its association with SMAD3 are both involved in mechanisms that mediate EMT [34][39]. Moreover, SMYD3 methylates two different receptor tyrosine kinases: vascular endothelial growth factor receptor 1 (VEGFR1), thereby potentiating angiogenesis through ligand-dependent autophosphorylation and increasing VEGFR1 kinase activity [17], and human epidermal growth factor receptor 2 (HER2), thereby enhancing HER2 homodimerization and subsequent autophosphorylation [18].
Overall, SMYD3 is a versatile coregulator of multiple oncogenic pathways, affecting processes associated with gene expression and protein transactivation through which it integrates cellular signals and promotes cancer development.
Several lines of evidence support the hypothesis that SMYD3 upregulation has a key role in tumorigenesis and cancer development in a number of human malignancies. Most of the studies performed to date showed a correlation between SMYD3 overexpression and cell growth in cancer settings. Knockdown of SMYD3 has been reported to decrease cell proliferation in a wide variety of cancers [6][7][10][15][22][23][32][35][36][40][41][42], while its overexpression promotes cell growth, transformation, and reduces apoptosis sensitivity [6][43]. Based on these findings, small-molecule SMYD3is have been generated, and several studies showed that SMYD3 inhibition affects cellular proliferation [9][27][29].
However, a recent paper by Thomenius et al. has called into question the role of SMYD3 in cancer cell growth by showing that SMYD3i or SMYD3 KO with the novel CRISPR/Cas9 technology failed to impair cell proliferation of hundreds of cancer cell lines of different origin and genetic background. Based on these findings, the authors concluded that SMYD3 is not required for autonomous proliferation of cancer cells in vitro [28].
In vivo studies on mice models seem to support SMYD3 involvement in tumorigenesis [9][11][15]. In a previous paper by our group, the expression and activity of SMYD3 were evaluated in a preclinical model of CRC, i.e., APCMin/+ mice treated with the carcinogen azoxymethane (AOM), and found to be strongly upregulated throughout tumorigenesis at both the mRNA and the protein levels, along with its downstream targets [9]. In another report, Mazur et al. showed that SMYD3 deficiency inhibits tumor development in mouse models of pancreatic ductal adenocarcinoma and lung adenocarcinoma, demonstrating that SMYD3 activity promotes the formation of RAS-driven carcinomas [15]. In line with these data, it has been shown that SMYD3 is required in mice for the development of chemically induced liver and colon carcinogenesis [11].
In this light, in-depth studies of the functional role of SMYD3 and its overexpression in cancer are instrumental to elucidate the mechanism by which it regulates oncogenic progression.
2.1 Role of SMYD3 in Controlling Cell Cycle Progression
It has been reported that overexpressed SMYD3 regulates cell growth by causing an acceleration of cancer cell division through modulation of the cell cycle [9][14][37][42][44]. Previous work by our group showed that SMYD3 affects cell cycle progression, revealing that its pharmacological inhibition by the novel small-molecule compound BCI-121 effectively reduces CRC cell proliferation by arresting cell cycle at the S/G2 boundary. This suggests the potential involvement of SMYD3 in the S/G2 checkpoint and hence in cell cycle deregulation, one of the critical steps in cancer development [9]. How SMYD3 affects cell cycle checkpoints is currently under study. Due to its ability to modulate chromatin accessibility, it can promote the transcription of several cell cycle-related genes. Sarris et al. demonstrated that SMYD3 occupies regulatory regions of genes involved in cell cycle control, such as CCNA2, CCNE1, CCND1, PCNA, IGFBP1, MYC, and CTNNB1, and showed that their expression decreases in the liver and colon of carcinogen-treated SMYD3-KO mice [11]. In addition, SMYD3 is recruited on the hTERT promoter, where it is required for the maintenance of H3K4 trimethylation in CRC and HCC cells. As a result, it supports the occupancy of the trans-activators c-MYC and Sp1, thereby promoting hTERT expression and its telomerase activity, which is essential for replicative immortality [33]. Interestingly, SMYD3 knockdown was shown to induce G2-phase arrest in GC cell lines, along with decreased expression of CDK1 and Cyclin B, which drives entry into mitosis, and higher levels of ATM and its downstream factors p53, CHK2, p21, and phosphorylated-Cdc25C, which contributes to G2 checkpoint control [42]. Furthermore, in HCC and esophageal squamous cell carcinoma, SMYD3 overexpression was associated with the expression of retinoblastoma protein-interacting zinc finger 1 (RIZ1) [22][41], which has a role in the G2/M checkpoint and is downregulated in several types of human cancers [45]. Specifically, high levels of SMYD3 were found to be associated with RIZ1 promoter hypermethylation, resulting in decreased RIZ1 mRNA expression [22][41].
Taken together, these reports define a critical role for SMYD3 in cell cycle progression. In particular, SMYD3 seems to be involved in S phase transition control and in the subsequent G2 checkpoint, which is a crucial cell cycle “timeout” in which DNA is checked for errors before mitosis can begin. Cancer cells usually ignore cell cycle checkpoints, which can lead to gain-of-function alterations in oncogenes and/or loss-of-function alterations in tumor suppressor genes [46]. In the event of DNA damage, proliferation is stopped and cells activate the DNA repair machinery to correct the error(s) or, when the damage cannot be repaired, they undergo cell death. Indeed, the G2 checkpoint is an essential safeguard mechanism to maintain genomic stability during cell cycle progression and is thus a critical process for cancer initiation and development.
2.2 Role of SMYD3 in DNA Damage: From Tumorigenesis to Cancer Progression
SMYD3 was found overexpressed in liver tumors in mice treated with diethylnitrosamine (DEN) as a model for HCC, and in colon tumors in mice treated with dimethylhydrazine/dextran sodium sulfate (DMH/DSS) and APCMin/+ mice treated with the carcinogen AOM as a model for CRC [9][11]. Knocking out SMYD3 dramatically reduced the tumor formation capacity induced by these carcinogens, as shown by a decrease in the number and size of tumor foci in the colon and liver compared to wild-type mice [11]. In line with recent observations on SMYD3 involvement in cancer development [28], no spontaneous liver tumor formation was detected in mice constitutively overexpressing SMYD3 in hepatocytes and no differences in tumor foci numbers were observed between wild-type and SMYD3-overexpressing mice after DEN treatment [11]. Remarkably, it has been shown that SMYD3 is required for the compensatory proliferation of cells that escaped apoptosis caused by DEN-induced and DMH/DSS-induced DNA damage [11]. These events, which are involved in carcinogenesis, could be an effect of SMYD3-mediated transcriptional regulation of cancer-related genes, such as MYC and CTNNB1, and components of the IL6-JAK-STAT3 pathway [11]. This evidence suggests that SMYD3 may play a signal-dependent role in promoting gastrointestinal (GI) cancer formation and development in response to genotoxic stress.
Interestingly, we have recently delved into the role of SMYD3 in maintaining genome integrity in a GI cancer context. Since SMYD3 regulates several key cancer-associated proteins through direct interaction, we carried out an in silico peptide screening with the aim of identifying new SMYD3 interactors to better characterize its involvement in cancer progression [47]. We found that SMYD3 directly binds to ATM, CHK2, and BRCA2, which are important sensors and effectors of homologous recombination (HR), a specific signaling cascade that is required for DNA DSB repair. Our results showed that high levels of SMYD3 are required for DNA restoration after the induction of DSBs. Specifically, SMYD3 promotes the formation of HR complexes during DDR by interacting with ATM. This propagates the signal cascade through CHK2 and BRCA2, thereby promoting the recruitment of RAD51 on DNA lesions. Moreover, new findings were obtained based on the identification of a new SMYD3 genetic variant (p.Arg265His) in a BC high-risk family [47]. This SMYD3-R265H mutant protein, which is predicted to be deleterious and was also found in a dataset of patients with CRC [48][49], shows a stronger interaction with ATM and localizes at DSBs like the wild-type form but is not able to interact with CHK2 and BRCA2. This prevents the recruitment of the DNA repair complex on damage sites, suggesting that this variant may play a dominant-negative role [47].
These new findings reveal an important role for SMYD3 in DNA repair and are supported by another study in which SMYD3 was linked to HR. In this paper, the authors focused on SMYD3-mediated modulation of the expression of genes related to DNA damage response and showed how SMYD3 influences DNA restoration by analyzing long recovery times [50]. In addition, as previously reported, it has been found that SMYD3 genetic ablation upregulates ATM and its downstream signaling cascade, thus suggesting that SMYD3 may influence cell cycle progression through an ATM-dependent mechanism [42].
These findings reveal that SMYD3 has an important protective function for cancer cells. Indeed, cancer cells display a high incidence of activated oncogenes resulting in uncontrolled pathways that sustain unlimited cell proliferation. This leads to error accumulation and DNA replication stress, which could compromise cell division and cancer progression. In this scenario, SMYD3 overexpression reinforces DNA damage response in cells with intrinsic/genotoxic stress and hence promotes cancer progression, suggesting that it may also alter cell sensitivity to genotoxic cancer therapy.
3. Clinical Impact of SMYD3 Inhibition for New Therapeutic Strategies in GI Cancers
Cancer cells are strictly dependent on DNA repair for survival and proliferation. Indeed, the DNA repair deficiency that occurs in some cancers results in the activation of alternative repair pathways [51]. These are mediated by PARP1 activity, which plays an important role as a sensor protein recognizing both single-strand breaks and DSBs and recruits DDR factors to the region around DNA lesions, thereby priming the activation of specific DNA repair cascades [52][53]. Recently, studies carried out to devise novel therapeutic strategies for cancer treatment have been focusing on DDR deficiencies with the aim of achieving synthetic lethality, which refers to the induction of cell death through combined deficiencies in the expression or activity of two genes, whereas the perturbation of either gene alone is viable [54][55]. These deficiencies can be the result of genetic mutations, epigenetic alterations, or the activity of specific inhibitors. Targeting the rescue DNA repair pathway in cancer cells carrying DDR deficiencies has been recently shown to be an effective strategy for several cancer types, including BRCA1/2-deficient cancers, where the use of PARP inhibitors (PARPi) is a model example of synthetic lethality [54]. The use of PARPi has entered clinical practice following FDA approval for the treatment of OvCas, BCs, and PCs harboring defects in HR genes [56], which define a BRCAness phenotype. The first PARPi, Olaparib, was approved for BRCA-mutated OvCa in 2014 [57]. It has subsequently been included in clinical trials for various types of GI cancers, including esophageal cancer, recurrent or metastatic GC, advanced PC, and CRC, often in combination with radio- and chemotherapy [58], since previous studies had shown a synergistic response to the combined treatment with PARPi and specific chemotherapeutics in GI cancers [59][60][61].
Currently, pharmaceutical companies are also directing their attention to other DDR and cell cycle checkpoint factors, including CHK1/2, ATM, ATR, DNA-PKcs, and RAD51, in order to develop cancer treatments to be used either alone or in combination with other anticancer drugs [58]. In addition, recent studies in preclinical models have shown the potential of the pharmacological inhibition of a DDR factor in a setting where another DDR factor is functionally defective [61].
Previous data from our group suggested the possible activation of compensatory DNA repair signals after inhibition of SMYD3 to impair HR repair. Since the activation of alternative DNA repair mechanisms requires PARP1 activity, we hypothesized that combined inhibition of SMYD3 and PARP1 with specific compounds could act as a synthetic lethality strategy. Specifically, we selected a tumor subset that is HR-proficient, and therefore addicted to high levels of SMYD3, as a candidate for this new therapeutic strategy based on the impairment of HR repair response with a specific SMYD3i to make the tumor sensitive to PARPi. Based on our hypothesis, the combined treatment would alter cancer cell ability to restore DNA damage and therefore cause cell death. Our results confirmed its potential, with the combined use of SMYD3i and PARPi showing a cytotoxic effect in CRC and PC cell lines [47]. Based on these findings, a synthetic lethality approach may be extended to a fraction of human tumors determined to be eligible for this therapeutic strategy. Eligibility could be assessed by evaluating a recently defined biomarker named HR deficiency (HRD) score, which determines the HR repair response status by analyzing specific standardized parameters [62]. Tumors with a low HRD score (meaning they are HR repair proficient) and high SMYD3 expression are expected to be the best candidates for the combined treatment (Figure 1). Based on an analysis of the PanCanAtlas dataset, we found that 41,2% of CRC tumors (from the COAD-READ dataset) with high SMYD3 mRNA levels have a low HRD score. Intriguingly, we also found that CRCs displayed mutual exclusivity of SMYD3 overexpression and genetic alterations of major HR genes that were previously shown to be correlated with a higher HRD score [47]. In the same study, we extended this analysis to another type of GI cancer by assessing a PC tumor dataset (PAAD). Our findings revealed that about 11% of total CRCs and PCs could be eligible for the combined treatment with SMYD3i and PARPi [47].
Altogether, this evidence supports the potential of a novel therapeutic strategy using a combination of SMYD3i and PARPi for HR-proficient tumors expressing high levels of SMYD3 (Figure 1). Based on our data on CRC and PC cell lines and on patient information from the above-mentioned datasets, this approach may be particularly effective in GI cancers.
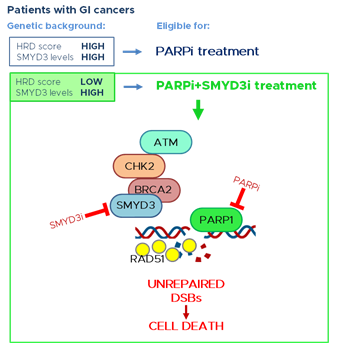
Figure 1. PARP inhibition is a promising therapeutic strategy for tumors with a high HRD score, thereby having a deficiency in the HR repair response. New evidence supports the potential of a novel therapeutic strategy for GI cancers with a low HRD score (HR proficient) and high levels of SMYD3. This strategy is based on a synthetic lethality approach consisting of the combined treatment with SMYD3 and PARP inhibitors. This would alter GI cancer cell ability to restore DNA damage and therefore lead to cell death. GI: gastrointestinal; SMYD3i: SMYD3 inhibitor; PARPi: PARP inhibitor; DSBs: double-strand breaks.
4. Conclusions and Future Directions
SMYD3 mediates the progression of several cancer types by regulating oncogenic mechanisms and signaling pathways in different ways. Here, we focused on SMYD3 involvement in tumors related to the GI compartment, where its altered expression has been found linked to cancer initiation, progression, and aggressiveness. SMYD3 can promote cancer by co-regulating the activation of major cancer-related pathways and can act as a critical driver in tumorigenesis. As for its role in cancer progression, SMYD3 has been described as a core promoter of cell cycle regulation that is involved in phase transition and allows cancer cells to bypass signals of cell cycle arrest, thereby contributing to uncontrolled proliferation. In addition, it has a protective role against genotoxic stress, which is critical for cancer development. SMYD3 contributes to the restoration of damaged DNA in cancer cells and therefore enables unperturbed cell division. Thus, SMYD3 appears as a genetic guardian of DNA damage checkpoint dynamics, driving cell cycle phase transition and promoting genomic protection of cancer cells.
Based on these findings, SMYD3 is emerging as an important target for drug discovery. Further studies are needed not only to gain a full comprehension of SMYD3-mediated mechanisms promoting cancer progression but also to gather stronger evidence in support of the effectiveness of novel therapeutic strategies based on the use of SMYD3i in specific patient subsets. Moreover, future studies will have to focus on the design of novel inhibitors suitable for cancer patients, with the aim of making SMYD3 a druggable target in clinical practice. Indeed, combining currently used DNA-damaging drugs with compounds that target DNA damage checkpoints can lead cancer cells to overcome repair mechanisms and cell cycle arrest, thereby undergoing cell death. In this light, a thorough understanding of the effects of SMYD3 inhibition may help to devise more selective and efficient pharmacological interventions for GI cancer patients in the clinical setting. In particular, it may allow to improve current therapies by combining them with SMYD3i to sensitize GI cancers expressing high levels of SMYD3.
References
- Bottino, C.; Peserico, A.; Simone, C.; Caretti, G. SMYD3: An Oncogenic Driver Targeting Epigenetic Regulation and Signaling Pathways. Cancers (Basel) 2020, 12, doi:10.3390/cancers12010142.
- Foreman, K.W.; Brown, M.; Park, F.; Emtage, S.; Harriss, J.; Das, C.; Zhu, L.; Crew, A.; Arnold, L.; Shaaban, S.; et al. Structural and Functional Profiling of the Human Histone Methyltransferase SMYDPLoS One 2011, 6, e22290, doi:10.1371/journal.pone.0022290.
- Zhang, Y.; Li, C.; Yang, Z. Is MYND Domain-Mediated Assembly of SMYD3 Complexes Involved in Calcium Dependent Signaling? Front Mol Biosci 2019, 6, 121, doi:10.3389/fmolb.2019.00121.
- Sirinupong, N.; Brunzelle, J.; Doko, E.; Yang, Z. Structural Insights into the Autoinhibition and Posttranslational Activation of Histone Methyltransferase SmyDJ Mol Biol 2011, 406, 149–159, doi:10.1016/j.jmb.2010.12.014.
- Brown, M.A.; Foreman, K.; Harriss, J.; Das, C.; Zhu, L.; Edwards, M.; Shaaban, S.; Tucker, H. C-Terminal Domain of SMYD3 Serves as a Unique HSP90-Regulated Motif in Oncogenesis. Oncotarget 2015, 6, 4005–4019.
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 Encodes a Histone Methyltransferase Involved in the Proliferation of Cancer Cells. Nat Cell Biol 2004, 6, 731–740, doi:10.1038/ncb1151.
- Cock-Rada, A.M.; Medjkane, S.; Janski, N.; Yousfi, N.; Perichon, M.; Chaussepied, M.; Chluba, J.; Langsley, G.; Weitzman, J.B. SMYD3 Promotes Cancer Invasion by Epigenetic Upregulation of the Metalloproteinase MMP-Cancer Res 2012, 72, 810–820, doi:10.1158/0008-5472.CAN-11-1052.
- Luo, X.-G.; Zhang, C.-L.; Zhao, W.-W.; Liu, Z.-P.; Liu, L.; Mu, A.; Guo, S.; Wang, N.; Zhou, H.; Zhang, T.-C. Histone Methyltransferase SMYD3 Promotes MRTF-A-Mediated Transactivation of MYL9 and Migration of MCF-7 Breast Cancer Cells. Cancer Lett 2014, 344, 129–137, doi:10.1016/j.canlet.2013.10.026.
- Peserico, A.; Germani, A.; Sanese, P.; Barbosa, A.J.; Di Virgilio, V.; Fittipaldi, R.; Fabini, E.; Bertucci, C.; Varchi, G.; Moyer, M.P.; et al. A SMYD3 Small-Molecule Inhibitor Impairing Cancer Cell Growth. J Cell Physiol 2015, 230, 2447–2460, doi:10.1002/jcp.24975.
- Van Aller, G.S.; Reynoird, N.; Barbash, O.; Huddleston, M.; Liu, S.; Zmoos, A.-F.; McDevitt, P.; Sinnamon, R.; Le, B.; Mas, G.; et al. Smyd3 Regulates Cancer Cell Phenotypes and Catalyzes Histone H4 Lysine 5 Methylation. Epigenetics 2012, 7, 340–343, doi:10.4161/epi.19506.
- Sarris, M.E.; Moulos, P.; Haroniti, A.; Giakountis, A.; Talianidis, I. Smyd3 Is a Transcriptional Potentiator of Multiple Cancer-Promoting Genes and Required for Liver and Colon Cancer Development. Cancer Cell 2016, 29, 354–366, doi:10.1016/j.ccell.2016.01.013.
- Kim, H.; Heo, K.; Kim, J.H.; Kim, K.; Choi, J.; An, W. Requirement of Histone Methyltransferase SMYD3 for Estrogen Receptor-Mediated Transcription. J Biol Chem 2009, 284, 19867–19877, doi:10.1074/jbc.M109.021485.
- Kim, J.-M.; Kim, K.; Schmidt, T.; Punj, V.; Tucker, H.; Rice, J.C.; Ulmer, T.S.; An, W. Cooperation between SMYD3 and PC4 Drives a Distinct Transcriptional Program in Cancer Cells. Nucleic Acids Res 2015, 43, 8868–8883, doi:10.1093/nar/gkv874.
- Tsai, C.-H.; Chen, Y.-J.; Yu, C.-J.; Tzeng, S.-R.; Wu, I.-C.; Kuo, W.-H.; Lin, M.-C.; Chan, N.-L.; Wu, K.-J.; Teng, S.-C. SMYD3-Mediated H2A.Z.1 Methylation Promotes Cell Cycle and Cancer Proliferation. Cancer Res 2016, 76, 6043–6053, doi:10.1158/0008-5472.CAN-16-0500.
- Mazur, P.K.; Reynoird, N.; Khatri, P.; Jansen, P.W.T.C.; Wilkinson, A.W.; Liu, S.; Barbash, O.; Van Aller, G.S.; Huddleston, M.; Dhanak, D.; et al. SMYD3 Links Lysine Methylation of MAP3K2 to Ras-Driven Cancer. Nature 2014, 510, 283–287, doi:10.1038/nature13320.
- Yoshioka, Y.; Suzuki, T.; Matsuo, Y.; Nakakido, M.; Tsurita, G.; Simone, C.; Watanabe, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. SMYD3-Mediated Lysine Methylation in the PH Domain Is Critical for Activation of AKTOncotarget 2016, 7, 75023–75037, doi:10.18632/oncotarget.11898.
- Kunizaki, M.; Hamamoto, R.; Silva, F.P.; Yamaguchi, K.; Nagayasu, T.; Shibuya, M.; Nakamura, Y.; Furukawa, Y. The Lysine 831 of Vascular Endothelial Growth Factor Receptor 1 Is a Novel Target of Methylation by SMYDCancer Res 2007, 67, 10759–10765, doi:10.1158/0008-5472.CAN-07-1132.
- Yoshioka, Y.; Suzuki, T.; Matsuo, Y.; Tsurita, G.; Watanabe, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. Protein Lysine Methyltransferase SMYD3 Is Involved in Tumorigenesis through Regulation of HER2 Homodimerization. Cancer Med 2017, 6, 1665–1672, doi:10.1002/cam4.1099.
- Mouse Genome Informatics. Available Online: URL (Http://Www.Informatics.Jax.Org/Allele/Key/571089).
- Zhou, Z.; Jiang, H.; Tu, K.; Yu, W.; Zhang, J.; Hu, Z.; Zhang, H.; Hao, D.; Huang, P.; Wang, J.; et al. ANKHD1 Is Required for SMYD3 to Promote Tumor Metastasis in Hepatocellular Carcinoma. J Exp Clin Cancer Res 2019, 38, 18, doi:10.1186/s13046-018-1011-0.
- Wang, Y.; Xie, B.-H.; Lin, W.-H.; Huang, Y.-H.; Ni, J.-Y.; Hu, J.; Cui, W.; Zhou, J.; Shen, L.; Xu, L.-F.; et al. Amplification of SMYD3 Promotes Tumorigenicity and Intrahepatic Metastasis of Hepatocellular Carcinoma via Upregulation of CDK2 and MMPOncogene 2019, 38, 4948–4961, doi:10.1038/s41388-019-0766-x.
- Dong, S.-W.; Zhang, H.; Wang, B.-L.; Sun, P.; Wang, Y.-G.; Zhang, P. Effect of the Downregulation of SMYD3 Expression by RNAi on RIZ1 Expression and Proliferation of Esophageal Squamous Cell Carcinoma. Oncol Rep 2014, 32, 1064–1070, doi:10.3892/or.2014.3307.
- Zhu, Y.; Zhu, M.-X.; Zhang, X.-D.; Xu, X.-E.; Wu, Z.-Y.; Liao, L.-D.; Li, L.-Y.; Xie, Y.-M.; Wu, J.-Y.; Zou, H.-Y.; et al. SMYD3 Stimulates EZR and LOXL2 Transcription to Enhance Proliferation, Migration, and Invasion in Esophageal Squamous Cell Carcinoma. Hum Pathol 2016, 52, 153–163, doi:10.1016/j.humpath.2016.01.012.
- Liu, X.; Zheng, Z.; Chen, C.; Guo, S.; Liao, Z.; Li, Y.; Zhu, Y.; Zou, H.; Wu, J.; Xie, W.; et al. Network Analyses Elucidate the Role of SMYD3 in Esophageal Squamous Cell Carcinoma. FEBS Open Bio 2017, 7, 1111–1125, doi:10.1002/2211-5463.12251.
- Zhu, C.-L.; Huang, Q. Overexpression of the SMYD3 Promotes Proliferation, Migration, and Invasion of Pancreatic Cancer. Dig Dis Sci 2020, 65, 489–499, doi:10.1007/s10620-019-05797-y.
- Fabini, E.; Manoni, E.; Ferroni, C.; Rio, A.D.; Bartolini, M. Small-Molecule Inhibitors of Lysine Methyltransferases SMYD2 and SMYD3: Current Trends. Future Med Chem 2019, 11, 901–921, doi:10.4155/fmc-2018-0380.
- Mitchell, L.H.; Boriack-Sjodin, P.A.; Smith, S.; Thomenius, M.; Rioux, N.; Munchhof, M.; Mills, J.E.; Klaus, C.; Totman, J.; Riera, T.V.; et al. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med Chem Lett 2016, 7, 134–138, doi:10.1021/acsmedchemlett.5b00272.
- Thomenius, M.J.; Totman, J.; Harvey, D.; Mitchell, L.H.; Riera, T.V.; Cosmopoulos, K.; Grassian, A.R.; Klaus, C.; Foley, M.; Admirand, E.A.; et al. Small Molecule Inhibitors and CRISPR/Cas9 Mutagenesis Demonstrate That SMYD2 and SMYD3 Activity Are Dispensable for Autonomous Cancer Cell Proliferation. PLoS One 2018, 13, e0197372, doi:10.1371/journal.pone.0197372.
- Van Aller, G.S.; Graves, A.P.; Elkins, P.A.; Bonnette, W.G.; McDevitt, P.J.; Zappacosta, F.; Annan, R.S.; Dean, T.W.; Su, D.-S.; Carpenter, C.L.; et al. Structure-Based Design of a Novel SMYD3 Inhibitor That Bridges the SAM-and MEKK2-Binding Pockets. Structure 2016, 24, 774–781, doi:10.1016/j.str.2016.03.010.
- Talibov, V.O.; Fabini, E.; FitzGerald, E.A.; Tedesco, D.; Cederfeldt, D.; Talu, M.J.; Rachman, M.M.; Mihalic, F.; Manoni, E.; Naldi, M.; et al. Discovery of an Allosteric Ligand Binding Site in SMYD3 Lysine Methyltransferase. ChemBioChem 2021, 22, 1597–1608, doi:10.1002/cbic.202000736.
- Alshiraihi, I.M.; Jarrell, D.K.; Arhouma, Z.; Hassell, K.N.; Montgomery, J.; Padilla, A.; Ibrahim, H.M.; Crans, D.C.; Kato, T.A.; Brown, M.A. In Silico/In Vitro Hit-to-Lead Methodology Yields SMYD3 Inhibitor That Eliminates Unrestrained Proliferation of Breast Carcinoma Cells. Int J Mol Sci 2020, 21, E9549, doi:10.3390/ijms21249549.
- Hamamoto, R.; Silva, F.P.; Tsuge, M.; Nishidate, T.; Katagiri, T.; Nakamura, Y.; Furukawa, Y. Enhanced SMYD3 Expression Is Essential for the Growth of Breast Cancer Cells. Cancer Sci 2006, 97, 113–118, doi:10.1111/j.1349-7006.2006.00146.x.
- Liu, C.; Fang, X.; Ge, Z.; Jalink, M.; Kyo, S.; Björkholm, M.; Gruber, A.; Sjöberg, J.; Xu, D. The Telomerase Reverse Transcriptase (HTERT) Gene Is a Direct Target of the Histone Methyltransferase SMYDCancer Res 2007, 67, 2626–2631, doi:10.1158/0008-5472.CAN-06-4126.
- Fenizia, C.; Bottino, C.; Corbetta, S.; Fittipaldi, R.; Floris, P.; Gaudenzi, G.; Carra, S.; Cotelli, F.; Vitale, G.; Caretti, G. SMYD3 Promotes the Epithelial-Mesenchymal Transition in Breast Cancer. Nucleic Acids Res 2019, 47, 1278–1293, doi:10.1093/nar/gky1221.
- Wang, S.; Luo, X.; Shen, J.; Zou, J.; Lu, Y.; Xi, T. Knockdown of SMYD3 by RNA Interference Inhibits Cervical Carcinoma Cell Growth and Invasion in Vitro. BMB Rep 2008, 41, 294–299, doi:10.5483/bmbrep.2008.41.4.294.
- Zou, J.-N.; Wang, S.-Z.; Yang, J.-S.; Luo, X.-G.; Xie, J.-H.; Xi, T. Knockdown of SMYD3 by RNA Interference Down-Regulates c-Met Expression and Inhibits Cells Migration and Invasion Induced by HGF. Cancer Letters 2009, 280, 78–85, doi:10.1016/j.canlet.2009.02.015.
- Ren, T.; Wang, J.; He, Y.; Xu, C.; Wang, S.; Xi, T. Effects of SMYD3 Over-Expression on Cell Cycle Acceleration and Cell Proliferation in MDA-MB-231 Human Breast Cancer Cells. Med Oncol 2011, 28 Suppl 1, S91-98, doi:10.1007/s12032-010-9718-6.
- Wang, T.; Wu, H.; Liu, S.; Lei, Z.; Qin, Z.; Wen, L.; Liu, K.; Wang, X.; Guo, Y.; Liu, Q.; et al. SMYD3 Controls a Wnt-Responsive Epigenetic Switch for ASCL2 Activation and Cancer Stem Cell Maintenance. Cancer Lett 2018, 430, 11–24, doi:10.1016/j.canlet.2018.05.003.
- Zhang, L.; Jin, Y.; Yang, H.; Li, Y.; Wang, C.; Shi, Y.; Wang, Y. SMYD3 Promotes Epithelial Ovarian Cancer Metastasis by Downregulating P53 Protein Stability and Promoting P53 Ubiquitination. Carcinogenesis 2019, 40, 1492–1503, doi:10.1093/carcin/bgz078.
- Luo, X.-G.; Ding, Y.; Zhou, Q.-F.; Ye, L.; Wang, S.-Z.; Xi, T. SET and MYND Domain-Containing Protein 3 Decreases Sensitivity to Dexamethasone and Stimulates Cell Adhesion and Migration in NIH3T3 Cells. J Biosci Bioeng 2007, 103, 444–450, doi:10.1263/jbb.103.444.
- Chen, L.-B.; Xu, J.-Y.; Yang, Z.; Wang, G.-B. Silencing SMYD3 in Hepatoma Demethylates RIZI Promoter Induces Apoptosis and Inhibits Cell Proliferation and Migration. World J Gastroenterol 2007, 13, 5718–5724, doi:10.3748/wjg.v13.i43.5718.
- Wang, L.; Wang, Q.-T.; Liu, Y.-P.; Dong, Q.-Q.; Hu, H.-J.; Miao, Z.; Li, S.; Liu, Y.; Zhou, H.; Zhang, T.-C.; et al. ATM Signaling Pathway Is Implicated in the SMYD3-Mediated Proliferation and Migration of Gastric Cancer Cells. J Gastric Cancer 2017, 17, 295–305, doi:10.5230/jgc.2017.17.e33.
- Luo, X.-G.; Xi, T.; Guo, S.; Liu, Z.-P.; Wang, N.; Jiang, Y.; Zhang, T.-C. Effects of SMYD3 Overexpression on Transformation, Serum Dependence, and Apoptosis Sensitivity in NIH3T3 Cells. IUBMB Life 2009, 61, 679–684, doi:10.1002/iub.216.
- Jiang, Y.; Lyu, T.; Che, X.; Jia, N.; Li, Q.; Feng, W. Overexpression of SMYD3 in Ovarian Cancer Is Associated with Ovarian Cancer Proliferation and Apoptosis via Methylating H3K4 and H4KJ Cancer 2019, 10, 4072–4084, doi:10.7150/jca.29861.
- Jiang, G.L.; Huang, S. Adenovirus Expressing RIZ1 in Tumor Suppressor Gene Therapy of Microsatellite-Unstable Colorectal Cancers. Cancer Res 2001, 61, 1796–1798.
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer Drug Resistance: An Evolving Paradigm. Nat Rev Cancer 2013, 13, 714–726, doi:10.1038/nrc3599.
- Sanese, P.; Fasano, C.; Buscemi, G.; Bottino, C.; Corbetta, S.; Fabini, E.; Silvestri, V.; Valentini, V.; Disciglio, V.; Forte, G.; et al. Targeting SMYD3 to Sensitize Homologous Recombination-Proficient Tumors to PARP-Mediated Synthetic Lethality. iScience 2020, 23, 101604, doi:10.1016/j.isci.2020.101604.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov 2012, 2, 401–404, doi:10.1158/2159-8290.CD-12-0095.
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Science Signaling 2013, 6, pl1, doi:10.1126/scisignal.2004088.
- Chen, Y.-J.; Tsai, C.-H.; Wang, P.-Y.; Teng, S.-C. SMYD3 Promotes Homologous Recombination via Regulation of H3K4-Mediated Gene Expression. Scientific Reports 2017, 7, 3842, doi:10.1038/s41598-017-03385-6.
- Klinakis, A.; Karagiannis, D.; Rampias, T. Targeting DNA Repair in Cancer: Current State and Novel Approaches. Cell Mol Life Sci 2020, 77, 677–703, doi:10.1007/s00018-019-03299-8.
- Gibson, B.A.; Kraus, W.L. New Insights into the Molecular and Cellular Functions of Poly(ADP-Ribose) and PARPs. Nat Rev Mol Cell Biol 2012, 13, 411–424, doi:10.1038/nrm3376.
- Pascal, J.M. The Comings and Goings of PARP-1 in Response to DNA Damage. DNA Repair (Amst) 2018, 71, 177–182, doi:10.1016/j.dnarep.2018.08.022.
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158, doi:10.1126/science.aam7344.
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic Lethality and Cancer. Nature Reviews Genetics 2017, 18, 613–623, doi:10.1038/nrg.2017.47.
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A Decade of Clinical Development of PARP Inhibitors in Perspective. Ann Oncol 2019, 30, 1437–1447, doi:10.1093/annonc/mdz192.
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res 2015, 21, 4257–4261, doi:10.1158/1078-0432.CCR-15-0887.
- Hosoya, N.; Miyagawa, K. Targeting DNA Damage Response in Cancer Therapy. Cancer Sci 2014, 105, 370–388, doi:10.1111/cas.12366.
- Reilly, N.M.; Novara, L.; Di Nicolantonio, F.; Bardelli, A. Exploiting DNA Repair Defects in Colorectal Cancer. Mol Oncol 2019, 13, 681–700, doi:10.1002/1878-0261.12467.
- Bang, Y.-J.; Im, S.-A.; Lee, K.-W.; Cho, J.Y.; Song, E.-K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J Clin Oncol 2015, 33, 3858–3865, doi:10.1200/JCO.2014.60.0320.
- Mauri, G.; Arena, S.; Siena, S.; Bardelli, A.; Sartore-Bianchi, A. The DNA Damage Response Pathway as a Land of Therapeutic Opportunities for Colorectal Cancer. Ann Oncol 2020, 31, 1135–1147, doi:10.1016/j.annonc.2020.05.027.
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep 2018, 23, 239-254.e6, doi:10.1016/j.celrep.2018.03.076.


