COVID-19 symptoms are highly heterogeneous, and the patient may be asymptomatic or may present with mild to severe or fatal symptoms. Factors, such as age, sex, and comorbidities, are key determinants of illness severity and progression. Aging is accompanied by multiple deficiencies in interferon production by dendritic cells or macrophages in response to viral infections, resulting in dysregulation of inflammatory immune responses and excess oxidative stress. Age-related dysregulation of immune function may cause a more obvious pathophysiological response to SARS-CoV-2 infection in elderly patients and may accelerate the risk of biological aging, even after recovery. Resveratrol is a potent antioxidant with antiviral activity.
1. Introduction
The emerging novel coronavirus disease 2019 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, has burdened health systems around the world. Identifying potential risk factors and predicting disease progression may be very useful for healthcare professionals to effectively classify patients, provide personalized treatment, monitor clinical progress, and allocate appropriate resources at all levels of care to reduce morbidity and mortality
[1].
Elderly people are very sensitive to viral infections; thus, disease severity and subsequent mortality are both higher in them than in younger people. COVID-19 in elderly people generally results in a high viral load, impaired virus clearance, high oxidative stress, increased inflammation, and inflammatory cytokine production
[2]. In severe cases, SARS-CoV-2 induces the release of cytokines and chemokines, such as interleukin (IL), interferon (IFN), and tumor necrosis factor (TNF) at a higher rate than normal (known as a “cytokine storm”), which can cause cytopathic effects and lead to organ failure
[3]. Although treatment to reduce the basic reproductive potential (R0) of the virus is essential, a search for specific treatments should not be ignored. The treatment of severely ill patients is supportive of this new type of coronavirus. However, effective therapies that are inexpensive, non-toxic, and readily available are urgently needed. Resveratrol (3, 5, 4/-trihydroxytrans-stilbene; RSV) is a naturally-occurring polyphenol compound found in grapes, red wine, blackberries, and groundnuts. RSV has received attention because of its therapeutic effects. In this regard, it has antioxidant, antitumor, antiviral, and free radical scavenging properties; thus, it can be regard as a potential adjuvant therapy
[4][5]. RSV modulates the inflammatory response in a pleiotropic manner, can scavenge free radicals such as superoxide, and may interfere with infections by altering numerous cell defense pathways.
[3][6].
2. Hyperinflammation in COVID-19
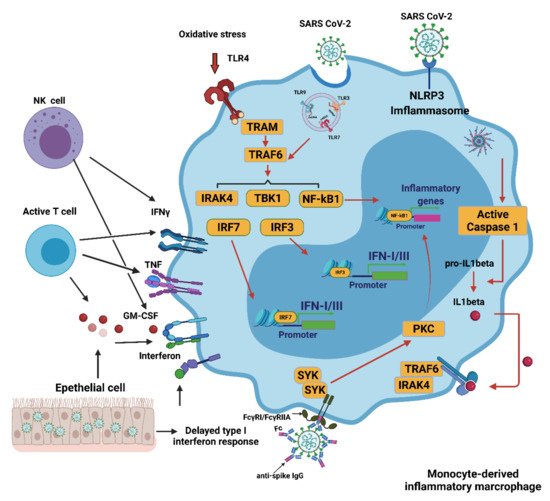
In COVID-19, multiple mechanisms can lead to the excessive activation of monocyte-derived macrophages. The delayed production of type I interferon (IFN I) leads to aggravated cellular pathological injury and the increased synthesis and secretion of mononuclear chemoattractants from alveolar epithelial cells, which leads to the continuous recruitment of blood monocytes to the lungs
[7]. Infection with SARS-CoV-2 may trigger the JAK/STAT inflammatory pathway, which causes the differentiation of monocytes into pro-inflammatory macrophages and contributes to the development of a cytokine storm
[8]. Recruitment and switch of monocyte-derived macrophages can be achieved by secreting granulocyte-macrophages colony stimulant factor (GM-CSF), tumor necrosis factor (TNF), and interferon-γ (IFNγ) from activated natural killer (NK) cells and T cells
[9]. The recognition of oxidized products such as oxidized phospholipids (OxPLs) in the infected lung can further facilitate the activation of macrophages to secrete pro-inflammatory cytokines and chemokines through the toll-like receptor 4 (TLR4)-tumor necrosis factor receptor associated factor 6 (TRAF6)-nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) pathway
[10]. The single-stranded RNA virus can be detected by LRT7 resulting in its activation. SARS-CoV-2 can cause neutrophils to release neutrophil extracellular traps (NETs). However, the NETs triggered by SARS-CoV-2 depend on angiotensin-converting enzyme 2 (ACE2), various proteases, peptidylarginine deiminase 4 (PAD4), and viral replication. NETs released by neutrophils can also promote lung epithelial cell death by increasing oxidative stress
[11]. IFN I can induce the expression of ACE2 for SARS-CoV-2 entry into the cytoplasm of activated macrophages and further activate the Nod-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasome, leading to the secretion of either mature IL-1β, IL-18, or both. IL-1 β may amplify the activation of monocyte-derived macrophages in an autocrine or paracrine manner but may also reduce the production of IFN I in the infected lung
[12]. In addition, anti-SARS-CoV-2 IgG antibodies are produced in B cells, but they can induce an antibody-dependent enhancement mechanism (ADE). This occurs with both sub- and non-neutralizing antiviral antibodies, resulting in the severity of the inflammation and oxidative stress being exacerbated. In vitro modeling of ADE has attributed the increased pathogenesis to viral entry of the Fcγ receptor (FcγR). The combination of the FcγR and the IgG immune complex of the anti-spike protein can significantly increase the number of inflammatory macrophages
[13]. These activated macrophages contribute to the oxidative stress and cytokine storm seen in a COVID-19 infection. These activated macrophages then release large quantities of pro-inflammatory cytokines and chemokines
[14] (
Figure 1).
Figure 1. Activation of monocyte-derived macrophages in COVID-19. Several mechanisms induce the excessive activation of monocytes/macrophages during a SARS-CoV-2 infection. The delayed production of IFN I results in the continued conscription of circulating monocytes into the pulmonary parenchyma. Activated NK and T cells also favor infiltrating cells derived from monocytes. Virus detection may trigger the activation of TLR7 as a result of the recognition of the viral single-stranded RNA. IFN I increases the expression of the SARS-CoV-2 entry receptor ACE2, allowing the virus to enter macrophages and activate cytoplasmic inflammation through NLRP3. The combination of the immune complex containing the anti-spike protein IgG with the Fcγ receptor (FcγR) on activated macrophages further promotes aberrant viral entry and causes an inflammatory cascade. CCL: CC-chemokine ligand; CXCL10: CXC-chemokine ligand 10; ISG: interferon-stimulated gene; ITAM: immune 06receptor tyrosine-based activation motif; TRAM: TRIF-related adaptor molecule; NK, natural killer. Figure generated with Biorender (
https://biorender.com/ accessed on 6 September 2021).
3. SARS-CoV-2 Infection and Oxidative Stress
Recent studies have suggested that oxidative stress is a key factor in the pathogenesis of COVID-19
[15][16]. Binding of SARS-CoV-2 to host cell membrane ACE2 facilitates virus entry into the host cell, thereby leading to a reduction in bioavailable ACE2. Because of the protective role of ACE2, a decrease in its level is associated with subsequent undesirable clinical phenotypes through activation of the NLRP3 inflammasome cascade
[17]. The high ratio of neutrophils to lymphocytes in critically ill patients with COVID-19 is associated with excessive levels of reactive oxygen species (ROS)
[18]. This oxidative stress can also be linked to ACE2. Whenever there is a dysfunction in ACE2 or when its levels are reduced due to SARS-CoV-2 infection, the overexpression of angiotensin II (Ang II) becomes a powerful pro-oxidant system in vessels and mononuclear cells
[19]. Ang II has been demonstrated to bind to the type 1 angiotensin receptor (AT1R) and activate nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)
[20][21][22][23]. NOX activation contributes to the excessive production of ROS, such as superoxide radical anions (O
2•−) and hydrogen peroxide (H
2O
2). This AngII mediated activation of AT1R signaling turns on NOX and induces oxidative stress and inflammation, resulting in severe COVID-19 symptoms
[24].
The ROS generated by NOX reduce the bio-available nitric oxide and cause inflammation, vasoconstriction, a redox imbalance, and endothelial dysfunction
[25][26]. ROS also contributes to the overexpression of NF-kB and thioredoxin interaction/inhibitory protein (TXNIP). NF-κB enhances the expression of NLRP3, pro-IL-1β, and pro-IL-18. TXNIP regulates the assembly of the NLRP3 inflammasome, which contributes to severe inflammation
[14]. Therefore, the cytopathic effects of SARS-CoV-2 may result in pyroptosis
[27][28], an inflammatory form of cell death elicited by inflammasomes, which leads to the breakdown of gasdermin D (GSDMD) and activation of inactive cytokines such as IL-18 and IL-1β. Violi et al. demonstrated that NADPH oxidase-2 (NOX-2) is amplified in hospitalized COVID-19 patients
[29].
The release of iron from red blood cells into the blood stream is another source of ROS in COVID-19 patients. This pathogenesis occurs because SARS-CoV-2 can damage hemoglobin (Hb) in the RBCs, thereby eliciting the release of free Fe(III) ions from affected heme into the circulation
[30]. This in turn increases serum ferritin levels
[31]. Therefore, SARS-CoV-2-mediated hemoglobinopathy and a dysregulation of iron metabolism contribute to ferroptosis, oxidative stress, lipid and protein peroxidation, and mitochondrial injury
[32].
4. Innate Immune Response to COVID-19
4.1. Innate Immune Response
The innate immune response to respiratory pathogens is a highly ordered process, involving several different layers of defense
[33]. In respiratory infections, respiratory epithelial cells, mast cells, and macrophages can detect invasive pathogens
[34]. PAMPs combine with host sensor cell membrane pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I) receptors, and adaptive molecules, resulting in an immune response tailored to the pathogen
[35]. PAMPs trigger an association between PRRs and adaptor molecules, leading to an immune reaction. Endosomal PRRs include toll-like receptors (TLRs) 3, 7, and 8, which recognize extracellular PAMPs such as viral RNA
[36]. Cytoplasmic RNA sensors such as RIG-I
[35] and MDA5 (melanoma differentiation-associated protein 5) can also bind to viral double-stranded RNA (dsRNA)
[37].
Intracellular DAMPs released from dead cells
[38] initiate PRRs in epithelial cells to trigger pro-inflammatory cytokine release
[39]. Thereafter, more effective mononuclear cells and T lymphocytes are attracted to further drive the inflammatory process.
Detecting coronavirus through RIG-I and other PRRs triggers an obvious innate immune response, which would effectively limit viral replication
[39]. First, IFN-I (IFN-α and IFN-β) or IFN-III cytokines are released to promote adequate eradication of the virus
[40]. Secreted IFN activates IFN-stimulated genes (ISGs) that exert direct antiviral properties and recruit effective antiviral immune cells such as myeloid cells
[41].
4.2. Interferons (IFNs), Proinflammatory Cytokines, and Oxidative Stress
IFNs are important antiviral cytokines
[40] and almost all nucleated cells respond to IFN I. The response to IFN III is restricted to the barrier of the respiratory tract or digestive tract
[42], but long-term treatment with IFN-III in viral infections may be harmful
[43].
An earlier report revealed elevated inflammatory cytokines and IFNs in bronchial alveolar lavage (BAL) in patients with severe COVID-19
[43]. Although robust ISG induction by IFNs was noted, no obvious modulation of IFN-I was observed in BAL cells
[44]. Replication of SARS-CoV-2 also resulted in decreased levels of IFN-I and IFN-II in a cellular model and in postmortem lung tissue of patients with COVID-19
[45]. It has also been reported that in ex vivo human lung tissue explants, SARS-CoV-2 did not significantly induce IFN-I
[46]. In patients severely affected by COVID-19, an elevated viral load has been reported to be associated with a decreased IFN-I response
[47] (
Figure 2).
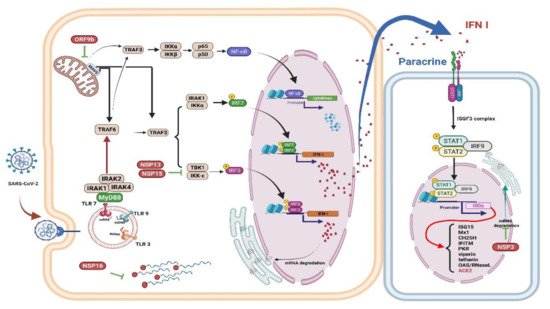
Figure 2. Immune evasion by SARS-CoV-2. The schematic shows how SARS-CoV-2 viral proteins are able to inhibit various immune processes such as pathogen recognition, IFN production and signaling, and ISGs
[48]. Studies have shown that each viral protein can block different key signaling cascades. Viral RNA can be assembled with guanosine and methylated at the 5’ end by SARS-CoV-2 non-structural proteins, allowing the virus to efficiently escape recognition of the viral dsRNA by the host cell sensor
[49]. Viral proteins inactivate key intermediaries in the IFN signaling cascade. A recent study showed that the SARS-CoV-2 ORF9b interacts with MAVS in mitochondria, resulting in a decrease in TRAF3 and TRAF6
[50]. The nsp13 and nsp15 proteins of SARS-CoV-2 interfere with TBK-1 signaling and activate IRF3
[50]. Another key virulence factor for SARS-CoV-2 is Nsp1, which inhibits the expression of the host gene; thus, it can effectively block the innate immune responses which could assist in the eradication of infection
[51]. The right panel shows that the viral protein Nsp3 blocks IFN signaling by reversible post-translational modification of ISG 15
[52]. In patients with severe COVID-19, a significant mitigation of IFN I response is associated with a clinically persistent viral load and increased oxidative stress and an inflammatory response
[47]. IFN: interferon; ISGs: interferon-stimulated genes; MAVS: mitochondrial antiviral signaling protein; nsp: nonstructural protein 13; TRAF: tumor necrosis factor receptor associated factor. Figure generated with Biorender (
https://biorender.com/accessed on 6 September 2021).
SARS-CoV-2 Nsp 1 (non-structural protein 1) can interfere with innate immune responses through RIG-I-dependent IFN-I/III production and virus removal.
[51]. Because replication of the coronavirus RNA occurs in a particular double-membrane vesicle (DMV) that separates the virus-related PAMP from the cytoplasmic RNA sensor, this compartmentalization also reduces the ability of the cytoplasmic sensor to detect virus replication
[53]. Finally, activating NF-κB significantly weakens IFN-I signaling, thus promoting viral replication
[54], indicates that a cross-regulatory function exists between the IFN-I/III and the NF-κB signaling cascades
[55]. NK cells are lymphocytes that play a significant role in the innate antiviral immune response. A recent in vivo study demonstrated that RSV activated NK cells and improved IFN-γ production and the cytotoxicity of NK cells, particularly in the presence of interleukin-2 (IL-2)
[56].
4.3. Neutrophils and NETs
Neutrophil extracellular traps (NETs) contributes to inflammation. When pathogens are present, circulating neutrophils release granular proteins and chromatin to form extracellular fibrous matrices called NETs. NETs bind to pathogens and neutralize them, undergoing a process of cell death called NETosis
[57]. Several enzymes participate in the formation of NETs, such as neutrophil elastase (NE)
[58][59], peptidyl arginine deiminase type 4 (PAD4)
[60], and gasdermin D
[61]; these enzymes facilitate leakage of the cell membrane and the expulsion of its DNA and related molecules (
Figure 3).
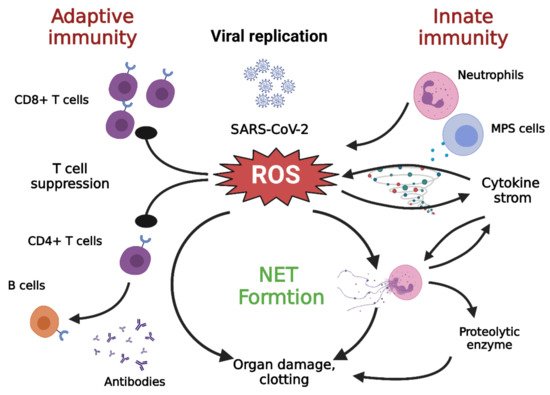
Figure 3. Cross talk among NETs, oxidative stress, and T cell deficiency
[28]. The immune pathogenesis of COVID-19 includes both innate and adaptive immune systems. As the virus escapes IFN-I/III surveillance, the long-term, large-scale replication of the virus is initiated in host pulmonary epithelial cells, monocyte/macrophages, and vascular endothelial cells. As a result, neutrophils and MPS cells are recruited in large numbers into inflamed tissues. T cells can kill infected cells and eventually eradicate the virus. In addition, CD4+ T cells are less effective in promoting B cells to generate neutralizing antibodies and develop a long-lasting immune response. DAMP: damage associated molecular pattern; IFN-I/III: interferon I/III; MPS, mononuclear phagocytic system; NETs, neutrophil extracellular traps; ROS: reactive oxygen species; TLR: toll-like receptor. Figure generated with Biorender (
https://biorender.com/accessed on 6 September 2021).
While NETs promote host immunity against pathogens, the collateral damage caused by persistent NET formation is exacerbated in many disease processes
[62]. NETs can facilitate macrophages to secrete IL1β, which further enhances the formation of NETs
[63][64]. Furthermore, excessive NET formation can trigger inflammatory reactions that promote the destruction of surrounding tissue and cause permanent damage to the lungs and vital organs
[65]. Furthermore, oxidative stress not only increases the formation of NETs but also contributes to the suppression of T lymphocytes
[28]. In addition, IL-1β can promote IL-6 expression, which in turn induces fibrin release and fibrinogen expression, while the virus-induced endothelial damage can expose tissue factors, all of which accentuate the coagulation pathway, which further aggravates fibrin deposition and clotting
[14].
4.4. Contribution of Monocytes/Macrophages to Oxidative Stress
Zhou et al. found significantly increased circulating CD14
+CD16
+ monocytes in hospitalized patients with COVID-19, a characteristic that was substantially prominent in patients with acute respiratory distress syndrome (ARDS). This finding suggests a certain degree of differentiation of monocytes to macrophages during SARS-CoV-2 infection. Circulating monocytes/macrophages express high levels of cytokines, such as IL-6, TNFα, and IL-10. This explains the relationship of these cells with oxidative stress, cytokine storm, and increased disease severity
[31].
Class II histocompatibility complexes, such as HLA-DR, are responsible for presenting antigens to CD4
+ T helper cells. The aberrant regulation of HLA-DR on monocytes and antigen-presenting cells, as is seen in cases of COVID-19, can result in systemic inflammatory conditions
[66].
5. Adaptive Immunity in COVID-19
An effective long-term antigen-specific immune response is key to controlling long-term viral infections and preventing viral persistence. Indeed, all individuals who have recovered from COVID-19 carry virus-specific CD4
+ T cells
[67], which is strongly indicative of a crucial role for the immune response in the successful elimination of SARS-CoV-2. This adaptive immunity is suppressed in elderly patients
[68] which could partially be because of defective monocyte-T cell crosstalk. Severe COVID-19 has also been associated with a higher frequency of depleted T cells
[69]. T lymphocyte depletion has been widely described as senescence
[70], further supporting the monocyte dysregulation, damage to adaptive immunity, and the severe COVID-19 seen in elderly patients (
Figure 3).
6. Aging in COVID-19
The number of deaths caused by the current COVID-19 pandemic is highly skewed toward older adults. Regardless of the country and stage of the outbreak, the death rate for COVID-19 increases exponentially with age
[71]. Immune senescence is thought to be a determining factor in SARS CoV-2 infections
[71][72].
Aging is associated with high levels of basal proinflammatory cytokines and acute phase proteins. The increased production of pro-inflammatory molecules and “inflamed aging” play a critical role in the development of the cytokine storm in patients with severe COVID-19
[73][74]. SARS-CoV-2 triggers a more potent NF-kB innate immune response in older animals than in younger animals, which results in an increased risk of ARDS
[55]. This effect is associated with age-related increase in ROS levels, which reflects a compensatory homeostasis reaction to support physiological cellular signaling in older people
[75]. The increase in ROS generation in older people further increases and reaches a threshold, causing NF-κB hyperactivity and inflammatory tissue injury
[76]. The delayed and inadequate IFN-1 response to SARS-CoV-2 allows sustained virus replication and heightened oxidative stress, which triggers an NF-κB-induced cytokine storm and excessive inflammation
[77][78]. Patients with COVID-19 have been shown to have upregulated NKG2A exhaustion markers in NK cells and CD8
+ cytotoxic T cells
[77]. In addition to the cytokine storm, SARS-CoV-2 can also directly damage multiple organs
[79] (
Table 1).
Table 1. Comparison of alterations in innate immune responses during aging (inflammaging) and the absence/reduction in type I IFN
[2][80].
|
Cell Type/Signaling
|
Aging
|
Absence/Reduction in Type I IFN
|
|
PRR activation & signaling
|
↑ High age-related basal PRR (TLR) activation leads to excessive pro-inflammatory cytokine production
↓ Post-PRR downstream signaling activation
|
↓ Recognition of intracellular pathogens
↑ Activity of Nlrp3 inflammasome
↓ ISGs expression signaling
↓Inducible nitric oxide synthase (iNOS)
|
|
Neutrophils
|
↑ Neutrophil influx through IL-17,
CXCL1, CXCL2
↓ Phagocytic function
↓ Signaling pathway
|
↑ Neutrophil recruitment by CXCL1 and CXCL2 production by monocytes
|
|
Monocyte/Macrophages
|
↑ IL-6 and TNF production
↓ Macrophage phagocytosis of
apoptotic neutrophils
↓ Alveolar macrophage affects repair of lung damage
|
↓ Inflammatory monocyte-derived macrophages (IM) recruitment
↑ Resident IM proliferation
↓ IM iNOS,
↑ Ly6Clo monocyte iNOS production
↓ IM TRAIL expression
↓ Macrophage phagocytosis and efferocytosis of apoptotic neutrophils
|
|
NK cells, Type I IFN
|
↓ Delayed type I IFN activation and production
↓ Cytotoxic early viral clearance
↑ Cytokine production and consequent lung damage
↑ NK cell apoptosis
|
↓ NK cell activation and IFN-production
↓ NK cell survival
|
|
Dendritic cells
|
↓ Functional capability
↓ DC maturation and migration to lymphoid organs affect T cell activation
|
↑ cDC2 subtype development
↓ Antiviral responses by lowering ISGs
↓ Migration function
|
↓: decrease; ↑: increase.
7. Resveratrol and COVID-19
7.1. Anti-Aging Interventions
While COVID-19 is frequently not lethal among the young, the death rate increases exponentially with age, especially in those with age-related illnesses. This suggests that age is probably a good predictor of mortality. At its deepest point, aging results from excessive abnormal cellular functioning, which explains why COVID-19 susceptibility is age-dependent and related to other age-related comorbidities. Anti-aging interventions, such as rapamycin, may slow the aging process and potentially reduce COVID-19 vulnerability
[81]. The natural anti-aging compound RSV has positive effects on lifetime, age-related illness, and maintenance of health. The different major mechanisms modulated by RSV can prevent or treat several age-related diseases. Thus, RSV appears to have a similar effect to rapamycin, even though it acts through a different mechanism
[82].
Aging physiology aims to understand how aging affects the body’s ability to withstand adverse events and how age-dependent molecular and cellular changes are involved in this process
[83]. The age-related exaggerated immune responses to COVID-19 that are characterized by a “cytokine storm”
[84] and intravascular coagulation
[85] may be attributed to the fact that the capacity of the human body to regulate its responses to threats is increasingly compromised during aging.
As many as 95% of COVID-19 victims have certain aging-related conditions, the prevalence of which increases exponentially with age
[86]. Thus, a potential measure to preventing or treating COVID-19 could be to utilize the methods that are generally suggested to alleviate the decline in age-dependent fitness and the associated development of age-related pathological processes.
7.2. Resveratrol as a Modulator of Antiviral Agent
Duck enteritis virus (DEV) is a double-stranded DNA virus belonging to the Alphaherpesvirinae sub-family of Herpesviridae
[87]. A previous study confirmed that RSV significantly reduces the mortality of ducklings infected with a virulent strain of DEV. Pathological symptoms and viral loads in blood and tissue were decreased effectively compared to the untreated group. In this study, a low concentration of RSV increased the production of IFN-α, IL-2, and IL-12, while these cytokine levels decreased by a high concentration of RSV
[88]. Rotavirus (RV) is a double-stranded RNA virus within the family Reoviridae. The gastroenteritis induced by RV, which is the foremost etiological agent for acute pediatric viral diarrhea worldwide, is a leading cause of death among children less than 5 years of age
[89]. RSV could alleviate diarrhea, inhibit the production of TNF-α, and increase IFN-γ levels after RV infection
[90]. Another study evaluating the efficacy of a nasal resveratrol/carboxymethyl-β-glucan (RSV/CMglucan) solution in infants with a common cold showed that it might have a positive impact on clinical outcomes
[91]. Middle Eastern respiratory syndrome (MERS) is a viral respiratory disease caused by the MERS coronavirus (MERS-CoV)
[92]. A study of MERS-infected Vero E6 cells showed that RSV significantly inhibited MERS-CoV infection and prolonged cellular survival after viral infection. Expression of the nucleocapsid (N) protein, which is essential for MERS-CoV replication, also decreased after RSV treatment. RSV also reduced apoptosis in the infected cells. Therefore, RSV may be useful as a treatment for MERS-CoV infections
[93].
ACE2 and TMPRSS2 both expressed in human cornea epithelial cells (HCEC). ACE2 expression is upregulated in HCECs, following stimulation with TNFα and IL-1β, and in inflamed corneas. RSV could attenuate the increased expression of ACE2 induced by TNFα in HCECs
[94]. With regard to the ACE2–spike interaction that occurs in COVID-19, molecular docking simulations provided evidence that RSV can bind to spike, ACE2, and the ACE2–spike complex with good affinity. Preliminary biochemical assays revealed a significant inhibitory effect of RSV on ACE2-spike binding
[95]. A recent mini-review found that the antiviral efficacy of RSV for a number of viruses, including the coronavirus. RSV can modulate the major pathways involved in the pathogenesis of SARS-CoV-2, including regulation of the renin-angiotensin system (RAS) and the expression of angiotensin-converting enzyme 2 (ACE2), stimulation of the immune system, and downregulation of pro-inflammatory cytokines release. RSV can stimulate the SIRT1 and p53 signaling pathways and increase cytotoxic T lymphocytes and NK cells. In addition, RSV treatment has been shown to be a fetal hemoglobin stimulator and a potent antioxidant that traps ROS
[96].
Various natural products have been found to be potential anti-COVID-19 agents. In this regard, it was suggested that RSV might be used alone or in combination with FDA-approved medications to treat COVID-19
[97]. The use of RSV in clinical practice is limited by low bioavailability following oral administration because of the pharmacokinetic and metabolic characteristics of the molecule. Therefore, topical administration through inhaled formulations, such as an aerosol, could allow the administration of sufficiently high concentrations of the compound through the airways, which are the entry route for SARS-CoV-2
[98][99]. RSV/CMglucan formulation may be appropriate for simultaneously volatilizing aerosols to treat patients with lower respiratory tract diseases
[95]. This could potentially suppress viral replication in the early stages of infection, reducing viral spread in the lower respiratory tract, leading to a reduction in the risk of infection transmission.
 +1 point
+1 point