Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Li Chuin Chong | + 1971 word(s) | 1971 | 2021-09-06 08:47:19 | | | |
| 2 | Conner Chen | Meta information modification | 1971 | 2021-09-22 04:06:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chong, L.C.; Khan, M.A. Alignment-Free Study of Viral Diversity. Encyclopedia. Available online: https://encyclopedia.pub/entry/14057 (accessed on 07 February 2026).
Chong LC, Khan MA. Alignment-Free Study of Viral Diversity. Encyclopedia. Available at: https://encyclopedia.pub/entry/14057. Accessed February 07, 2026.
Chong, Li Chuin, Mohammad Asif Khan. "Alignment-Free Study of Viral Diversity" Encyclopedia, https://encyclopedia.pub/entry/14057 (accessed February 07, 2026).
Chong, L.C., & Khan, M.A. (2021, September 10). Alignment-Free Study of Viral Diversity. In Encyclopedia. https://encyclopedia.pub/entry/14057
Chong, Li Chuin and Mohammad Asif Khan. "Alignment-Free Study of Viral Diversity." Encyclopedia. Web. 10 September, 2021.
Copy Citation
Viral sequence variation can expand the host repertoire, enhance the infection ability, and/or prevent the build-up of a long-term specific immunity by the host. The study of viral diversity is, thus, critical to understand sequence change and its implications for intervention strategies.
minimal set
alignment independent
alignment-free
sequence diversity
proteome
virus
UNIQmin
1. Introduction
Infectious diseases caused by viruses are a primary contributor to the global burden of death and disability [1][2]. The world is struggling against viral diseases, with billions of people afflicted annually, even impacting developed and developing countries with improved living conditions. The still ongoing coronavirus disease 2019 (COVID-19) pandemic, for instance, has threatened the global health systems, with a mortality of more than 3.5 million (https://coronavirus.jhu.edu; accessed on 30 May 2021) and no clear indication of when the disease will be brought under control.
Viral sequence variation, even of a single amino acid, can expand the host repertoire, as in the case of zoonotic viruses, or enhance the infection ability of a virus [3][4][5]. Sequence change can result in the evasion of host-established immunity and prevent the build-up of a long-term specific immunity [6]. This thus poses a challenge in the design of drugs and vaccines against viruses and can require a constant need to keep up with the evolving diversity [7][8][9][10]. The effect of viral antigenic diversity on vaccine efficacy and the need to keep up is well-demonstrated for influenza A virus (IAV), where annual re-formulation has been a recommendation by the World Health Organization (WHO) for decades [11]. Highly effective vaccines or drugs are still not publicly available for many notable viral diseases. The study of viral diversity is, thus, imperative in understanding sequence change and its implications for intervention strategies [9][12][13].
The rapid expansion in public sequence data provides an unprecedented opportunity to study viral adaptation and evolution. The National Center for Biotechnology Information (NCBI) public sequence databases consist of ~4.1 M nucleotide and ~10.9 M protein sequences (as of May 2021). Given the importance of viral diversity analyses, various sequence studies have been performed using alignment-dependent approaches [14][15][16][17] that focus on identifying and positioning corresponding regions of individual bases or amino acids. The utility of such an approach, however, is inversely proportional to the increase in sequence diversity, due to the corresponding decrease in conserved regions to anchor the alignment, particularly for highly diverse viruses [18]. This is further limiting when applied to the search for universal vaccine targets that capture the diversity of multiple subtypes or subgroups of a viral species, such as influenza A subtypes or human immunodeficiency virus 1 (HIV-1) clades. Moreover, when expanded to the genus rank, such conserved regions are typically non-existent [19]. Towards this, alignment-free or -independent approaches can offer an alternative to the study of sequence diversity. Such an approach can be defined as a method of quantifying sequence similarity or dissimilarity without the need to use dynamic programming or produce an alignment. Over the past decades, this has been implemented through a variety of methods, which can be mainly grouped into word frequency methods and those that do not resolve the sequence with fixed word-length segments [18].
Previously, Khan et al. (2005) described an alignment-independent method that performs an exhaustive search to determine the minimal set of sequences for a given dataset [20][21]. The minimal set herein refers to the smallest possible number of unique sequences required to represent the diversity inherent in the entire repertoire of overlapping k-mers encoded by all the unique sequences in the given dataset. Such dataset compression is possible through the removal of unique sequences, whose entire repertoire of overlapping k-mer(s) can be represented by other sequences, thus rendering them redundant to the collective pool of sequence diversity. Applied to a protein dataset for the study of amino acid substitutions, the complete set of peptides of a given k-mer length, encoded in the dataset, can be referred to as part of the viral peptidome [22]. The concept of a minimal sequence set is illustrated in Figure 1. Briefly, a given non-redundant (nr) dataset of unique sequences can possess a repertoire of k-mers that represents the inherent viral peptidome diversity (for the said k-mer length), which can be collectively covered by a smaller fraction of the unique sequences in the initial input dataset; this smaller fraction is termed as the minimal set. The study of a minimal set has thus far been reported for only two viruses, specifically at the species rank, namely Dengue virus (DENV) [20][21] and Influenza A virus (IAV) [23], which provided important insights into effective sequence diversity and evolution of the two viruses. Thus, this merits further exploration for other viruses, not just at the species rank, but at any rank of the viral taxonomy lineage, such as genus, family or even at the highest, superkingdom rank (all reported “Viruses”).

Figure 1. Definition of a minimal set. (A) Three unique dengue virus envelope protein sequences (QBE89964.1, QBE89970.1, and QBE89971.1) retrieved from the NCBI Entrez Protein Database are shown aligned (only a 25 amino acids fragment is shown for demonstration purposes). The consensus of the alignment is shown, with positions of variability indicated by the small case residues (variable amino acids A and L colored in red). (B) All the overlapping k-mers (9-mers in this example) are generated from each unique sequence. The 9-mers represent all the possible repertoire of viral peptidome diversity, relevant to the k-mer, present in the three sequences. Although these three sequences are each unique, they share identical 9-mers. The 9-mers shown in green color are those that are identical in all the three sequences, while those in cyan are identical between two of the sequences. The unique 9-mers are shown in black color with variable residues indicated in red. All the 9-mers in sequence QBE89964.1 have a match in at least one of the other two sequences; thus, the k-mer repertoire of this sequence can be collectively covered by the other two sequences, rendering the sequence QBE89964.1 as redundant. (C) Minimal set, requiring only two of the three unique protein sequences (QBE89970.1 and QBE89971.1) to capture the inherent k-mer repertoire of all the unique sequences in the initial dataset.
The reported algorithm by Khan et al. [20][21] is not scalable for large datasets at higher taxonomic lineages and thus requires optimization with regards to (i) the demand on the computational resource, (ii) optimality of the minimal set generated, and (iii) redundancies in the algorithm. To address these issues, we derived a novel algorithm that is significantly improved and scalable for massive datasets, such as all reported viral sequences in nature. The alignment-independent algorithm has also been implemented as a tool, UNIQmin, to allow for a user-specific search of the minimal set. The tool is open source and publicly available at https://github.com/ChongLC/MinimalSetofViralPeptidome-UNIQmin (accessed on 25 August 2021). We describe herein, the algorithm, the tool, its performance evaluation, and application to the study of viral diversity.
2. UNIQmin, An Alignment-Free Tool for the Study of Viral Sequence Diversity at Any Given Rank of Taxonomy Lineage
The idea of a minimal set, herein, is essentially a compression problem [24][25][26], applied to the study of viral protein sequence diversity, without incurring any loss of information in terms of the total peptidome repertoire (relevant to the k-mer of choice). Hence, the minimal set is the smallest fraction of the non-redundant protein sequences required to represent the complete peptidome repertoire present in the dataset (relevant to the k-mer). Thus, the minimal set can be considered to provide insight into the effective sequence diversity and evolution of the virus. The tool, UNIQmin, provides an alternative approach to analyze sequence diversity, commonly done using alignment-dependent approaches, which would be ineffective for the study of a diverse protein sequence dataset. A diverse dataset can be of a highly diverse virus species, such as Human immunodeficiency virus 1 (HIV-1), or spanning multiple species at higher taxonomic lineage ranks, such as genus or family. Thus, the approach herein may represent a major paradigm shift, as a direct enabler of novel applications in the study of sequence diversity and indirectly contributing to alignment-independent research. For example, the minimal set can be generated spatio-temporally to allow for comparative analyses of sequence diversity. Any decrease in compression relative to a referenced dataset of unique sequences would be indicative of an increase in the repertoire of novel k-mers in the collective pool of sequence diversity, while an increase would imply higher k-mer redundancy relative to the referenced dataset. Moreover, the minimal set can be subjected to further downstream analyses, coupled with alignment-dependent approaches, where applicable. It is expected that over time, the minimal set would show limited growth with increases in sequence data for a given taxonomy rank, assuming a balanced and sufficient sampling. This is because as the number of non-redundant sequences grows, viral variants exhibiting k-mers with novel sequence change, relative to the existing pool of sequence diversity, become less likely, and even if observed, such viral variants would be limited in number; unless the pool is perturbed by sequencing of previously unknown members of the taxonomy rank.
The UNIQmin algorithm (Figure 2) is a significant improvement from that of the Khan algorithm (KA) and one that offers the best combination of speed and optimality of the minimal set generated. It was observed to be 263-fold faster than ITERmin, a re-implementation of KA. UNIQmin achieved the speed-up performance by recognizing and taking advantage of the fact that singleton k-mers only occur once in the dataset. Thus, the sequences from which they originated are already candidates for the minimal set. This avoids not only the need to process those sequences but also eliminates the need to evaluate multi-occurring k-mers that are also present within the singleton-k-mer-harboring sequences. This pre-filtering, utilizing single-occurring k-mers, ended up capturing ~91.5% (5048 sequences) of the eventual 5519 dengue virus minimal set sequences (derived from a starting input dataset of 9800 nr DENV sequences), prior to the execution of the canonical core steps of the algorithm for the remaining sequences. More importantly, UNIQmin was scalable for application to big data. It was successfully applied to ~2.2 M nr protein sequences of all reported viruses, deduplicated from ~4.9 M downloaded sequences (retrieved as of November 2018), the number of which has now grown to ~9.9 M (as of May 2021), with ~3.3 M nr sequences. UNIQmin is expected to still manage well this increase in data, with a better speed-up performance utilizing machines with a larger number (>14) of CPU cores. We also demonstrated the utility of UNIQmin in compressing diverse datasets spanning lineage ranks, such as Flavivirus (genus) and Flaviviridae (family). The resulting minimal sets covered the proteomic diversity within (intra) and between (inter) the viral species members of the genus or family. Additionally, UNIQmin demonstrated a reasonable compression in bit-rate relative to other well-known tools (Gzip and AC), with compression directly proportional to sequence conservation within the dataset, without impacting the inherent diversity.
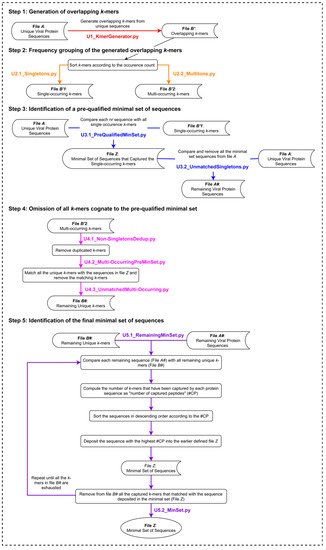
Figure 2. The UNIQmin algorithm. Abbreviation: nr, non-redundant.
3. Conclusions
UNIQmin enables the generation of a minimal set for a given sequence dataset of interest, without the need for sequence alignment. This alignment-independent approach allows one to select data that spans various ranks of the taxonomic lineage and provides the opportunity to ask relevant research questions with respect to effective sequence diversity, which would not be possible by the use of alignment-dependent approaches. Notably, it is also able to reduce big data size approximately by half, which is welcome, even though computing power is becoming cheap and more pervasive. This enables the exploration of big data compression prior to diversity or related analyses. This would allow for more efficient use of computer resources and would be a boon to those who have limited access to a big data computer infrastructure. The concept of a minimal set is generic and thus possibly applicable to both genomic and proteomic data of other pathogenic microorganisms of non-viral origin, which generally exhibit much larger data size, such as archaea (~7.0 M protein sequences as of May 2021), bacteria (~798 M), and Eukaryota (~108 M).
References
- Keni, R.; Alexander, A.; Nayak, P.G.; Mudgal, J.; Nandakumar, K. COVID-19: Emergence, Spread, Possible Treatments, and Global Burden. Front. Public Health 2020, 8, 216.
- GBD 2019 Diseases and Injuries Collaborator. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222.
- Steinhauer, D.A. Pathways to human adaptation. Nature 2013, 499, 412–413.
- Wendel, I.; Matrosovich, M.; Klenk, H.D. SnapShot: Evolution of Human Influenza A Viruses. Cell Host Microbe 2015, 17, 416–416.e1.
- Thakur, A.; Mikkelsen, H.; Jungersen, G. Intracellular Pathogens: Host Immunity and Microbial Persistence Strategies. J. Immunol. Res. 2019, 2019, 1356540.
- Volkov, I.; Pepin, K.M.; Lloyd-Smith, J.O.; Banavar, J.R.; Grenfell, B.T. Synthesizing within-host and population-level selective pressures on viral populations: The impact of adaptive immunity on viral immune escape. J. R. Soc. Interface 2010, 7, 1311–1318.
- Heiny, A.T.; Miotto, O.; Srinivasan, K.N.; Khan, A.M.; Zhang, G.L.; Brusic, V.; Tan, T.W.; August, J.T. Evolutionarily Conserved Protein Sequences of Influenza A Viruses, Avian and Human, as Vaccine Targets. PLoS ONE 2007, 2, e1190.
- Khan, A.M.; Miotto, O.; Nascimento, E.J.M.; Srinivasan, K.N.; Heiny, A.T.; Zhang, G.L.; Marques, E.; Tan, T.W.; Brusic, V.; Salmon, J.; et al. Conservation and Variability of Dengue Virus Proteins: Implications for Vaccine Design. PLOS Negl. Trop. Dis. 2008, 2, e272.
- Bingham, R.J.; Dykeman, E.C.; Twarock, R. RNA Virus Evolution via a Quasispecies-Based Model Reveals a Drug Target with a High Barrier to Resistance. Viruses 2017, 9, 347.
- Chong, L.C.; Khan, A.M. Identification of highly conserved, serotype-specific dengue virus sequences: Implications for vaccine design. BMC Genom. 2019, 20, 921.
- Regional Planning. Influenza Pandemic Plan. The Role of WHO and Guidelines for National and Regional Planning; World Health Organization: Geneva, Switzerland, 1999; pp. 1–66.
- Raman, H.S.A.; Tan, S.; August, J.T.; Khan, M.A. Dynamics of Influenza A (H5N1) virus protein sequence diversity. PeerJ 2020, 7, e7954.
- Hackbart, M.; Deng, X.; Baker, S.C. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl. Acad. Sci. USA 2020, 117, 8094–8103.
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and Evolution of the Global RNA Virome. mBio 2018, 9, e02329-18.
- Yang, O.O.; Ali, A.; Kasahara, N.; Faure-Kumar, E.; Bae, J.Y.; Picker, L.J.; Park, H. Short Conserved Sequences of HIV-1 Are Highly Immunogenic and Shift Immunodominance. J. Virol. 2015, 89, 1195–1204.
- Koo, Q.Y.; Khan, A.M.; Jung, K.-O.; Ramdas, S.; Miotto, O.; Tan, T.W.; Brusic, V.; Salmon, J.; August, J.T. Conservation and Variability of West Nile Virus Proteins. PLoS ONE 2009, 4, e5352.
- Yang, O.O. Candidate Vaccine Sequences to Represent Intra- and Inter-Clade HIV-1 Variation. PLoS ONE 2009, 4, e7388.
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 1–17.
- Chong, L.C.; Khan, A.M. Vaccine Target Discovery. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier BV: Amsterdam, The Netherlands, 2019; pp. 241–251.
- Khan, A.M. Mapping Targets of Immune Responses in Complete Dengue Viral Genomes. Master’s Thesis, National University of Singapore, Singapore, 2005; pp. 1–135.
- Khan, A.M.; Heiny, A.T.; Lee, K.X.; Srinivasan, K.N.; Tan, T.W.; August, J.T.; Brusic, V. Large-scale analysis of antigenic diversity of T-cell epitopes in dengue virus. BMC Bioinform. 2006, 7, S4.
- Özer, O.; Lenz, T.L. Unique Pathogen Peptidomes Facilitate Pathogen-Specific Selection and Specialization of MHC Alleles. Mol. Biol. Evolution. 2021, msab176.
- Heiny, A.T. The Antigenic Diversity Analysis of Complete Viral Genome of Influenza A Virus. Bachelor’s Thesis, National University of Singapore, Singapore, 2005; pp. 1–95.
- Hosseini, M.; Pratas, D.; Pinho, A.J. AC: A Compression Tool for Amino Acid Sequences. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 68–76.
- Hategan, A.; Tabus, I. Protein is compressible. In Proceedings of the 6th Nordic Signal Processing Symposium—NORSIG 2004, Espoo, Finland, 9–11 June 2004; pp. 192–195.
- Adjeroh, D.; Nan, F. On Compressibility of Protein Sequences. In Proceedings of the Data Compression Conference (DCC’06), Snowbird, UT, USA, 28–30 March 2006; pp. 422–434.
More
Information
Subjects:
Virology; Mathematical & Computational Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
22 Sep 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No