Immune check point blockade therapy has revolutionized the standard of cancer treatment and is credited with producing remarkable tumor remissions and increase in overall survival. This unprecedented clinical success however is feasible for a limited number of cancer patients due to resistance occurring before or during a course of immunotherapy, which is often associated with activation of oncogenic signaling pathways, co-inhibitory checkpoints upregulation or expansion of immunosuppressive regulatory T-cells (Tregs) in the tumor microenviroment (TME). Targeted therapy aiming to inactivate a signaling pathway such as the Mitogen Activated Protein Kinases (MAPKs) has recently received a lot of attention due to emerging data from preclinical studies indicating synergy with immune checkpoint blockade therapy. The dimeric transcription factor complex Activator Protein-1 (AP-1) is a group of proteins involved in a wide array of cell processes and a critical regulator of nuclear gene expression during T-cell activation. It is also one of the downstream targets of the MAPK signaling cascade.
1. Introduction
The 2018 Nobel Prize for Physiology or Medicine awarded to the pioneers of immune checkpoint research, James P. Allison and Tasuko Honjo, attests to the considerable advances achieved in the field of immunotherapy over the past decades. Immunotherapy, and more specifically immune checkpoint blockade therapy, represents a transforming event in the treatment of metastatic cancer since for the first time studies have shown that it can promote strong and durable tumor regression in some patients whose tumors have already metastasized
[1].
Immune checkpoint blockade (ICB) or immune check point inhibition therapy aims at the inhibition of the molecules collectively known as immune checkpoints, expressed mainly on the cells of the immune system. Immune checkpoints, whose physiological role is to maintain self-tolerance and restrict collateral damage in the tissues following immune responses, are frequently exploited by tumor cells to escape from the surveillance of the immune system which leads to immune suppression and promotion of tumor growth
[2]. In comparison to conventional treatment modalities in oncology (radiotherapy, chemotherapy etc.), ICB is innovative because it targets molecules expressed on the cells of the immune system, aiming to disrupt the tumor-derived immune suppression and reinvigorate the immune system to elicit a potent and oftentimes durable anti-tumor response. Monoclonal antibodies targeting immune checkpoints (e.g., CTLA-4 and PD-1) have so far received approval from the Food and Drug Administration (and other regulatory authorities) for demonstrating dramatic clinical responses in patients with metastatic cancers, leading to substantial improvement of their overall survival (OS) across diverse histological types and genetics of neoplastic diseases
[2][3].
Emerging evidence, however, suggests that ICB is beneficial only for a fraction of cancer patients, while it comes with a great cost of severe immune-related adverse events (irAEs) for a significant portion of those treated
[4]. Data from several clinical trials suggest that patients are stratified in the following three groups according to their resistance to ICB therapy: (1) responders, patients who respond to initial treatment and continue to respond, (2) non-responders, those that do not succeed in responding to ICB (primary resistance), and (3) those that initially respond but eventually develop disease progression (acquired or adaptive resistance)
[5][6]. Several factors have been identified within the TME, such as a genetic and epigenetic mutational load that controls neoantigen processing or presentation by the tumor cells, inhibitory checkpoint expression (e.g., PD-L1) and activation of oncogenic signaling pathways (tumor cell intrinsic mechanism) that suppress the therapeutic effects of ICB by disrupting the functions of tumor-specific cytotoxic T cells
[2][7]. In addition, tumor cell extrinsic mechanisms operating outside the TME such indole 2,3-dioxygenase (IDO) activity and the actions of immunosuppressive cells like T regulatory (Tregs) and myeloid-derived suppressor cells (MDSCs), is conducive towards an immunosuppressive environment, promoting tumor growth and resistance to ICB
[7].
A growing body of evidence indicate that activation of signaling pathways in various cancer types, such as the Mitogen-Activated Protein Kinases (MAPK)
[8], phosphatidylinositol 3-kinase (PI3K)
[9][10] and wnt/β-catenin
[11] can promote an immune-compromised tumor microenvironment, conferring resistance to immunotherapy. Preclinical evidence in various cancer types suggest that MAPK inhibition (MAPKi) can dramatically increase the efficacy of immunotherapy
[12][13][14][15][16][17] mainly via increased antigen presentation from tumor cells, augmented MHC-I expression, suppressed Treg expansion and increased proliferation and activation of tumor-infiltrating cytotoxic T-cells.
2. Activator Protein-1 (AP-1) Transcription Factors
It has been more than 30 years since the discovery of activator protein-1 (AP-1), described initially as a DNA-binding protein which recognized a DNA element found in the enhancer region of SV40 and the human Metallothionein IIA gene (
MT2A)
[18] and characterized by their ability to transactivate target genes upon phorbol ester stimulation (TPA)
[19]. These AP-1 transcription factors were later found to regulate a wide range of cellular processes spanning from cell proliferation and survival to tumor transformation, differentiation and apoptosis
[20]. AP-1 transcription factors are homo- or hetero-dimmer forming proteins that belong to a group of DNA binding proteins called Basic -Leucine Zipper domain (bZIP)
[21]. Dimerization between members of the AP-1 family occurs through a structure which is known as leucine zipper, comprised of a heptad of repeats of leucine residues along a α-helix, which can dimerize with another α-helix via formation of a coiled–coil structure with contacts between hydrophobic leucine zipper domain. Adjacent to the leucine zipper lies a basic DNA binding domain which is rich in basic amino acids and is responsible for DNA-binding in either 12-
O-tetradecanoylphorbol-13-acetate (TPA) response elements (5′-TGAG/CTCA-3′) or cAMP response elements (CRE, 5′-TGACGTCA-3′)
[20][22]. Members of the AP-1 protein family differ markedly in their potential to transactivate AP-1 responsive genes and their ability to form dimmers. For example, the Fos sub-family cannot homodimerize, but they can form stable heterodimers with Jun members
[23][24]. The Fos and Jun proteins have high transactivation potential, whereas others like JunB, JunD, Fra-1 and Fra-2 are weaker
[25]. Early studies using murine fibroblasts, substantiate the antagonistic nature of some AP-1 members against others. For instance, cJun transcriptional activity is attenuated by JunB and this is due to differences in their activation domains
[25][26]. Nevertheless, the current viewpoint suggests that the differential expression of AP-1 components and the cell context of their interactions determines the complex functions of AP-1 transcription factor
[21].
2.1. Regulation of Immune Response by AP-1 in Genetically Modified Mice
Early studies from transgenic animals implied a prominent role of AP-1 in the regulation of the immune system, arising from targeted overexpression of JunB in T lymphocytes
[27]. In these transgenic mice, elevated JunB levels caused upregulation of IL-4 expression, a cytokine which is known for facilitating the differentiation of Th2 cells while preventing the differentiation of Th1 cells. JunB binds to P1 element on
IL-4 promoter and synergizes with c-Maf to activate IL-4 luciferase reporter gene and JunB is also preferentially upregulated in developing Th2 cells. Collectively, this study suggests that JunB may contribute to the differentiation of naïve T-helper cells into Th2 during T cell development. In addition, in vivo data from transgenic mice expressing a mutant variant of cJun (JunAA), which is unable to sustain activation by JNK phosphorylation, reveal that even though T-cell activation and proliferation were not impaired in these mice, c-Jun N-terminal phosphorylation was required for efficient TCR- and TNFa (tumor necrosis factor-α)-induced thymocyte apoptosis, suggesting a role for cJun in thymocyte development
[28]. On the other hand, ectopic expression of the FosB2 gene in thymocytes causes aberrant development of T cells and thymic epithelial cells
[29].
2.2. AP-1 and T-Cell Activation
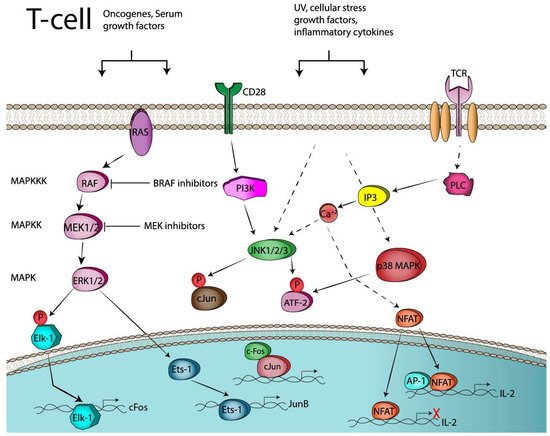
T-cell activation of naïve T-cells requires two signaling events
[30]. The initial signal, signal-1, is generated by interaction of a peptide antigen presented in association with an MHC molecule on the surface of an antigen presenting cell (APC). The supply of a subsequent co-stimulatory second signal (signal-2) which is delivered by interactions of CD28 on the T-cell with molecules on the APC is then required for full T-cell activation and production of cytokines (IL-2), proliferation and differentiation of effector cells
[31]. The signaling pathways that are activated by both signals (signal 1 and signal 2) are now well identified
[32] and they culminate in the activation of the enzyme phospholipase C (PLCγ), which cleaves the membrane lipid PI(4,5)P2 (phosphatidylinositol 4,5 bisphosphate) producing the second messengers IP3 and DAG (diacylglycerol). The first message, IP3, results in a rapid release of Ca
2+ from the ER and this will eventually lead to the activation of the transcription factor, nuclear factor of activated T-cells (NFAT) (
Figure 1). Ras, a small G protein which is dependent on GTP for activation, will initiate following TCR engagement the signaling cascade of MAPK which leads to phosphorylation of ERKs (Erk1/2) and the activation of Elk1 by phosphorylation. The latter translocates to the nucleus and binds to the promoter of
C-FOS, thus facilitating its transcription (
Figure 1)
[33]. An additional mechanism contributing to AP-1 activation from TCR engagement stems again from the MAPKs and the activation of the Jun-N-terminal kinase (JNKs)
[34] (
Figure 1). The JNKs, phosphorylate cJun, enhancing its transcriptional activity, leading to the formation of the AP-1 complex, Jun:Fos heterodimers, which are transcriptionally potent and thus bind and regulate target genes. cJun in several cancers (melanoma, colon cancer) is also induced through a MAPK independent mechanism, which involves cell–cell contacts and the adhesion molecule E-cadherin
[35]. Also, the CD28 pathways via PI3 kinase and acidic sphingomyelinase, can lead to the induction of AP-1
[36]. The effects of the AP-1 transcription factors, associated with the immune system, are largely mediated through combinatorial regulation with the NFAT, a calcium/calcineurin pathway-dependent transcription factor. The transcription factors of the NFAT family are key regulators of T cell activation
[37]. There are five members in the NFAT family members, of which NFAT1-4 (NFATc1-c4) are regulated by Ca
2+-calcineurin signaling. NFAT/AP-1 transcription factors bind cooperatively to composite DNA sites, where they participate in the formation of stable ternary complexes regulating the expression of target genes (
Figure 1). Composite DNA sites have been identified on the promoters of most of the cytokine genes, including
IL-2, IFN-g, TNF-a, GM-CSF, IL-4, FasL, CD25 and
NFAT/AP-1 where combinatorial regulation has been well documented
[38]. DNA-binding experiments have demonstrated that the NFAT/AP-1-binding complex contains predominantly cJun, c-Fos, JunB and Fra-1 proteins
[39].
Figure 1. Transcriptional and post-translational activation of AP-1 in T-cells. UV (Ultraviolet); CD28 (co-stimulation, signal-2), TCR (T-cell receptor, signal-1).
2.3. AP-1 and T-Cell Anergy or Exhaustion
T-cell “anergy” is an unresponsive state of T-cells in which T-cells are activated in the absence of a positive costimulatory signal, while T-cell “exhaustion” is referred to the state of CD8
+T cells that respond poorly because of prolonged antigen exposure during chronic viral infections or cancer
[40][41]. Some of the hallmarks of anergic T cells are the inhibition of proliferation and their inability to synthesize IL-2 in response to TCR engagement
[40]. Similarly, exhausted CD8
+T cells display a transcriptional program distinct from that of functional effector or memory CD8
+ T cells, characterized by the expression of inhibitory cell-surface receptors, including PD-1, LAG-3, TIM-3, TIGIT, and CTLA-4, and by impaired IL-2, TNF, and IFN-γ cytokine production. NFAT and AP-1 transcription factors synergistically play a central role in inducing hyporesponsive states, such as anergy and exhaustion
[42][43]. Exhausted cells exhibit low expression of AP-1 factors (Fos, Fosb, and Junb)
[44]. In addition, several lines of evidence suggest that cooperation of NFAT with AP-1 stimulates gene expression after immune response while absence of AP-1 leads to repression of the involved genes and to blockade of T-cell activation and proliferation, which eventually leads to T-cell anergy
[45][46]. NFAT, in the absence of cooperation with the transcription factor AP-1 (Fos-Jun), fosters a transcriptional program of genes associated with anergy and exhaustion in both CD4
+ and CD8 T cells, whereas, when AP-1 factors are present, NFAT drives expression of molecules such as cytokines that are involved in effector responses
[42].
Collectively, these findings indicate that the presence or absence of AP-1 in the transcriptional complexes with NFAT contributes greatly to the anergy/exhaustion phenotype of T-cells, reversal of which is an important clinical goal as demonstrated by the immune checkpoint blockade therapy. Therefore, targeting the AP-1:NFAT complexes might have therapeutic implications. Indeed in a recent study, Mognol et al. designed a FRET-based high-throughput screen to identify compounds that disrupt the NFAT:AP-1:DNA complex. They identified a small molecule, which disrupts the NFAT:AP-1 interaction at the composite antigen-receptor response element-2 site without affecting the binding of NFAT or AP-1 alone to DNA. This small molecule is capable of binding to DNA in a sequence-selective manner and inhibit the transcription of the IL2 gene and several other cyclosporin A-sensitive cytokine genes important for the effector immune response thus providing a proof-of-concept approach to target AP-1 transcription factors
[47].
3. AP-1 and Immune Checkpoint Regulation
According to the two-signal model, co-stimulatory molecules are responsible for sustained T-cell activation and effector T-cell function. Evidence for the two-signal model of T-cell activation was provided by the discovery of CD28 on T-cells, as the archetypal co-stimulatory molecule, which after binding to its ligand, it provides signal-2 stimulation, which along with TCR signal-1 is required for full T-cell activation. Subsequent discovery of CTLA-4 as an antagonistic molecule to the CD28 function, provided feedback for the negative stimulation of T-cells following activation and designated a group of molecules with similar function as co-inhibitory molecules. Ever since, the list of co-stimulatory and co-inhibitory molecules and their ligands is exponentially increasing along with potential clinical applications in patients with more than 10 cancer types, including metastatic melanoma, renal cell carcinoma, non-small-cell lung carcinoma (NSCLC), Hodgkin’s lymphoma and several others
[48][49][50]. Signaling downstream the immune checkpoint molecules is complex and has been reviewed in great extent elsewhere
[51], but there is evidence for the participation of several members of the AP-1 family
[51].
3.1. Co-Stimulatory Molecules and AP-1 Transcription Factors
3.1.1. CD28
CD28, a 44-kDa type I transmembrane glycoprotein, is constitutively present on the surface of naïve and activated T-cells
[52]. Stimulation via the CD28 pathway augments lymphokine production and proliferation in T cells while preventing induction of anergy
[51]. Inactivation of CD28 in vivo gives rise to immune compromised mice, characterized by impaired T-cell responses to antigen and defects in T-cell differentiation
[53]. The natural ligands for CD28 are the B7 family of adhesion proteins present on dendritic cells, activated B cells, and macrophages. Ligation of CD28 on T-cells with members of the B7 (CD80 or D86) family on antigen presenting cells provides signal-2. CD28 harbors a YMNM motif in his cytoplasmic tail through which it associates with the p85 subunit of PI3K, a common signaling intermediate, to initiate targeting of AKT (also known as protein kinase B (PKB)) that subsequently results in activation of several distal molecules. Co-stimulatory signals from CD28 ligation results in augmentation of downstream effector cascades, like the PI3 kinase, Ras and acidic sphingomyelinase
[51]. These pathways result in the activation of transcription factors such as NF-κB and AP-1, which mediate functional outcomes including IL-2 production and T cell survival
[54][39].
Insights for AP-1 involvement in CD28 pathway, comes from in vitro
[36][55] and in vivo
[56] studies, after CD28 co-stimulation. In part, stimulation of AP-1 activity is a result of JNK activation, which can occur through both TCR/CD3 and CD28 pathways, resulting in higher levels of JNK activity compared with signal 1 stimulation alone
[57]. In a study using T-cell blasts, investigators were able to observe the result of CD28 signaling in isolation without the TCR contribution. They found that CD28 costimulation induced AP-1 activity, which was dependent on PI3K and partly the acidic sphingomyelinase
[36]. Also c-jun mRNA induction was reported in T-cells after cross-linking of CD28. This CD28-dependent induction of c-jun expression requires protein tyrosine kinase activity but is Ca
2+ independent
[58]. Interestingly, CD28 also recruits the RAS guanine nucleotide exchange factor (GEF) RAS guanyl nucleotide-releasing protein (RASGRP) to the T cell-APC interface to induce activation of RAS and the downstream phosphorylation of AKT, JNKs and ERKs which are potent inducers of the AP-1 activity
[59]. Finally, CD28 engagement with B7 ligand, augments JNK signaling, which in turn regulates Elk-1 transactivation at the
c-FOS gene to promote AP-1 complexes which are important to
IL-2 gene expression
[60].
3.1.2. CD40/CD40L
The costimulatory receptor CD40 is a member of the tumor necrosis factor receptor (TNFR) superfamily. CD40 is expressed on dendritic cells, B cells, macrophages and also on non-hematopoietic cells, like endothelial cells and epithelial cells
[61]. CD40 binds to its ligand CD40L (or CD154), a type II transmembrane protein, which is transiently expressed primarily on the surface of activated B and T-cells and other non-immune cells
[62]. The wide expression pattern of CD40 and its ligand suggests a pivotal role in the regulation of immune-related processes
[62]. Ligation of CD40 results in clustering of CD40 and facilitates the recruitment of the TNFR associated factors (TRAFs) to the cytoplasmic domain of CD40
[63]. The TRAFs then activate several signaling pathways including the NFκB, the MAPKs, PI3K, as well as the phospholipase Cγ (PLCγ) pathway
[63]. TRAF2 and TRAF3 are involved in activation of the JNK pathway.
In human urothelial cells, engagement of the CD40 to membrane presented CD40L led to CD40-induced apoptosis involving TRAF3 and JNK/AP-1 activation
[64]. Furthermore, the murine
CD40L promoter contains NFAT binding motifs which require AP-1 cooperational binding for activation of transcription
[65]. In biliary epithelial cells, ligation of CD40 with the recombinant CD40L promoted Fas-dependent apoptosis and nuclear factor κB (NF-κB)/AP-1 signaling. Sustained activation of AP-1 in the absence of NF-κB signaling may be critical in determining the outcome of CD40 engagement
[66]. Besides, in B-lymphocytes, AP-1 proteins were found to control the mouse IL-6 expression after CD40 engagement, since mutations in the putative AP-1 (and C/EBP) binding sites on the murine
IL-6 promoter, abrogated promoter transcriptional activity. In the same study, CD40 stimulation led to phosphorylation of c-Jun on its activation domain, implicating CD40-mediated Jun kinase activation in the transcriptional regulation of IL-6 production
[67]. Another study, also in B cells, has demonstrated that stimulation through both CD40 and Toll-like receptor 7 (TLR7) enhanced the production of cytokines through increased JNK signaling and AP-1 activity. The increased level of active JNK in dual-stimulated cells was accompanied by an increase in the level of active AP-1 monomers cJun and cFos
[68]. Moreover, in cultured human fetal microglia cells, ligation of CD40 with soluble trimeric CD40L, results in augmentation of IL-8 (CXCL8) expression and this is mediated by activation of the ERK1/2 MAPK pathway. Gel shift analyses demonstrated that NFκB and AP-1, but not C/EBPβ mediate microglial CXCL8 production
[69].
3.2. Co-Inhibitory Molecules
3.2.1. PD-1
Programmed cell death protein-1 (PD-1), also known as CD279, is a type I transmembrane protein, whose expression is induced on activated immune cells such as T, B and NK cells. The major role of PD-1, in contrast to the CTLA-4, is to restrict the T-cell activation in peripheral tissues, to prevent from autoimmune disease and to maintain tolerance within the TME
[2]. The cytoplasmic domain of PD-1 contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM) which are speculated to have immunosuppressive properties
[70]. In the direct pathway, binding of PD-1 to its ligand, the B7 member PD-L1, strongly interferes with TCR/CD28 signal transduction and terminates ZAP70 and PI3K phosphorylation by recruiting the SHP1 and SHP2 phosphatases to its tyrosine phosphorylated ITIM and ITSM motifs
[71][72]. As a result, PD-1 abrogates cytokine production, causes cell cycle arrest and decreases transcription of the pro-survival factor Bcl-X
L [70]. Also, PD-1 inhibits RAS and, subsequently, its downstream targets ERK1 and ERK2 through an SHP1- and SHP2-independent mechanism
[51][70]. Because PD-1 is also highly expressed in Tregs where it regulates the development, maintenance, and function of induced regulatory T cells
[73], it makes it an ideal target for ICB, since PD-1 inhibition could also interfere with their function on the proliferation on the TME
[73].Therefore, PD-1 blockade could theoretically not only lead to enhancement of the activity of effector T cells and NK cells in the peripheral tissues but also to the restriction of the immunosuppressive action of Tregs in the TME
[2].
A study revealed that a major role of PD-1 is interfering with the AP-1 signaling generated from co-stimulatory cascades. PD-1 inhibits T-cells function by augmenting BATF expression. Accordingly, ectopic expression of BATF was sufficient to impair T cell proliferation and cytokine secretion, whereas BATF knockdown reduced PD-1 inhibition. Silencing BATF in T cells from individuals with chronic viremia rescued HIV-specific T cell function. PD-1, through BATF upregulation, activated a program of genes specific for exhausted CD8
+ T-cells, although the details of this molecular mechanism still remains elusive
[74]. In another study using a mouse model, tumor infiltrating T-cells exhibited high AP-1 activity and specifically expression of c-fos was shown to upregulate PD-1 in tumor infiltrating T-cells during tumor progression. Forced expression of c-fos in T-cells was associated with higher tumor burden, while T-cell specific depletion of c-fos led to reduction in tumor volume. C-fos was found to bind to the promoter region of
PD-1 and thus facilitates its expression. Therefore, blockade of c-fos mediated induction of PD-1 could be harnessed therapeutically to restore T-cells anti-tumor response
[75].
3.2.2. PD-L1
PD-L1 (CD274), one of the two ligands for PD-1, is a member of the B7 family of co-inhibitory molecules that negatively regulates T-cell immune responses. PD-L1 has a broad expression pattern and it is expressed in normal tissues (T and B cells, NK cells, macrophages, dendritic cells, epithelial cells, and vascular endothelial cells) and tumor cells
[76]. Specifically, ligation of PD-L1 of cancer cells to PD-1 expressed on T cells suppresses T-cell activation, proliferation, and induces T-cell apoptosis, which renders it an excellent target for ICB, using antibodies against PD-L1. The regulation of PD-L1 is complex and it occurs at the genetic, transcriptional and post-transcriptional levels
[77] and discussing it would be beyond the scope of this review.
In Hodgkin’s Lymphoma (HL), which is characterized by constitutive AP-1 activity
[78], AP-1 response elements were identified and demonstrate that cJun and JunB bind to an enhancer region of the
PD-L1 promoter, facilitating the PD-L1 expression along with Epstein-Barr virus (EBV) infection
[79]. Also in another EBV-associated tumor, the nasopharyngeal carcinoma (NPC), the EBV-induced latent membrane protein 1 (LMP1) and IFNγ, upregulated PD-L1 expression through AP-1, STAT3 and NF-κB pathways. These findings imply that blocking both the AP-1 oncogenic pathway and PD-1/PD-L1 checkpoints may be a promising therapeutic approach for EBV positive NPC tumors
[80].
In melanoma, PD-L1 is highly expressed in cell lines resistant to BRAF inhibitors (BRAFi). BRAFi-resistant cell lines developed dramatic activation in MAPK signaling pathways including extracellular signal-regulated kinase (ERK1/2), JNKs and p38. Increased activation of MAPK promotes PD-L1 expression in the BRAFi-resistant melanoma cells, associated with increased activity of c-Jun. Conversely, inhibition of c-Jun expression by siRNA led to significant decrease of PD-L1 in K028 resistant and parental M34 line, as well as near complete inhibition of PD-L1 expression in M34-resistant line. Thus, c-Jun promotes PD-L1 expression, which can be enhanced via cooperation of STAT3 in melanoma cells. These findings have important therapeutic implications for combining targeted treatment with immune modulation to improve antitumor responses and patient outcomes
[81].
In lung adenocarcinoma, MEK inhibition led to a marked reduction on surface PD-L1 levels in vitro, and similar results were seen after ERK2 gene silencing. Moreover, the
PD-L1 promoter was found to contain a functional AP-1 binding site, whose activity is abrogated by MEKi and cJun was bound to this AP-1 site. Overall, the study points to the seminal role of AP-1 in regulating PD-L1 expression, through MAPK upstream signaling
[82].
In a mouse model of chronic lymphocytic choriomeningitis virus (LCMV) infection, PD-1 blockade resulted in reinvigoration of exhausted T-cells (T
EX) but these changes were not accompanied by memory development and T
EX become again re-exhausted upon repeated antigen stimulation. The authors hypothesized that the genome-wide epigenetic landscape of T
EX may contribute to the lack of durable improvements after PD-L1 checkpoint blockade. Thus, they performed global chromatin landscape mapping using assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq). They found that anti-PD-L1 treatment caused transcriptional rewiring and reengagement of effector circuitry in the T
EX epigenetic landscape. Motif enrichment analysis of the few differentially accessible regions suggested that cells from anti-PD-L1—treated mice augmented activity of NF-κB, AP-1, and IRF family members but decreased activity of NFAT, Egr2, and Nur77. This study illustrates that AP-1 transcription factors constitute important players in the transcriptional circuitry of re-energized T
EX after anti-PD-L1 blockade.
[83].
 +1 point
+1 point