Identification of markers for the detection of tumor-initiating cells, first in leukemia by the group of John Dick (CD (cluster of differentiation) 34
+/CD38
−) and then in solid tumors including glioma (CD133
+, aldehyde dehydrogenase, ALDH1
+), breast (epithelial cell adhesion molecule, EpCAM
+/CD44
high/CD24
low, ALDH1
+), colorectal (CD133
+, EpCAM
high/CD44
+, ALDH1
+), head and neck squamous carcinoma (CD44
+), and other cancers as discussed elsewhere
[21], paved the way for isolating, enriching, and analyzing these tumorigenic cells in different tumor entities
[15][21][22]. These findings indicated that tumor-initiating cells, also called cancer stem cells (CSCs) possess two fundamental properties that make them different from other tumor cells; they have unlimited capacity to self-renew (e.g., divide asymmetrically to produce an identical copy of itself and more differentiated progeny cells) and differentiate to all cell populations present in the original tumors. These properties make these cells a root of tumor growth and recurrence and, thus, an important marker for tumor diagnosis, prognosis, and treatment, as well as critical targets for cancer therapy. Strong evidence is emerging to support the dynamic nature of tumor stemness which can be influenced by genetic alterations, epigenetic reprogramming and the tumor microenvironment
[13][23]. Although tumor cell heterogeneity displays a much higher level of complexity than can be explained by the hierarchical model, CSC populations remain critical targets and biomarkers for cancer treatments.
2. Cancer Stem Cells and 5Rs of Radiation Biology
Radiation therapy is one of the key anti-cancer treatment options along with surgery and chemotherapy
[6]. However, a high inter- and intratumoral variability in the phenotypical and functional properties of cancer cells remains the major obstacle to improving cancer survival rates, which is especially true for patients with locally advanced tumors
[15][24], and reliable biomarkers for patient stratification are of high demand
[25]. As of today, only few biological parameters are suggested as potential biomarkers for radiation oncology including tumor size, hypoxia, or positivity for human papilloma virus (HPV) for head and neck squamous cell carcinoma (HNSCC)
[11]. The biology-based patient stratification aims to select potential responders and non-responders to increase the probability of cancer cure by radiation therapy. It is based on the radiobiological concept of the “5Rs” which are repair, redistribution, repopulation, reoxygenation, and intrinsic radioresistance
[26][27].
The curative potential of radiotherapy depends on the scale and quality of DNA damage in the exposed tumor tissues. Ionizing radiation induces different types of DNA damage with DNA double-strand breaks (DSBs) as major lethal DNA lesions. If the levels of radiation-induced DSBs exceed the DNA repair capacity of tumor cells, this might result in cell cycle arrest, tumor cell senescence, and death. The cell fate decision following radiation exposure depends on the amount of critical DNA damage. A low level of DNA lesions triggers DNA repair mechanisms and DNA damage checkpoints, which arrest cell cycle progression in the presence of DNA damage and allow cells to repair DNA before returning to the proliferative pool
[28]. However, if the amount of DNA damage is unrepairable, the cells activate death programs
[28][29]. The first “R” refers to DNA repair as one of the key determinants of tumor cell survival after radiation therapy. The cellular response to DNA damage is a complex process including activation of the DNA damage response pathways and DNA repair mechanisms as reviewed elsewhere
[28][30]. In addition to direct DNA ionization, radiation-induced DNA damage is caused by highly chemically reactive species produced by radiolysis of water and by disruption of normal mitochondrial functions
[30]. These chemically active molecules, called reactive oxygen species (ROS) and reactive nitrogen species (RNS), induce damage of DNA, protein oxidation, and lipid peroxidation, and might cause cell senescence and death
[31]. These chemical species are regular by-products of cell metabolism and important second messengers
[31][32]. Under physiological conditions, the intracellular concentration of ROS is maintained by the scavenging system which includes, e.g., glutathione, thioredoxin, and enzymatic proteins thioredoxin reductase, dismutase, peroxidase, and catalase, all of which might also shield the cells against radiation-induced oxidative stress
[33].
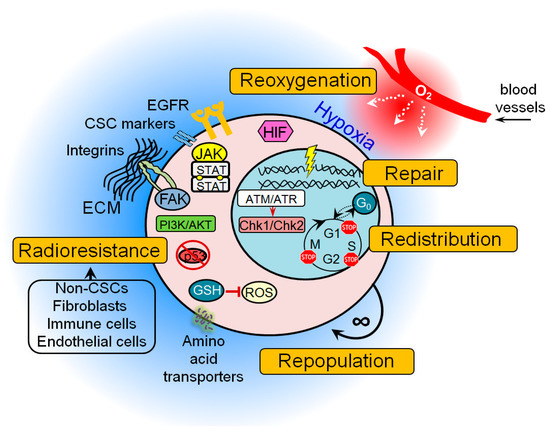
Accumulating preclinical evidence indicates that many of these protective mechanisms are activated in CSC populations possibly resulting in treatment resistance (Figure 1). These findings also suggest that signaling pathways that control DNA integrity and repair in CSCs may serve as a promising target in cancer therapy, as discussed in the next sections of this review.
Figure 1. The general mechanisms of cancer stem cell (CSC) radioresistance. Many protective mechanisms are activated in CSCs that result in treatment resistance. These include intrinsic determinants (activation of the pro-survival pathways, enhanced DNA repair capability, protection against oxidative stress, unlimited self-renewal potential, impaired cell cycle arrest, and apoptosis) and extrinsic determinants such as hypoxic microenvironment and a protective CSC niche consisting of the cellular components (e.g., non-CSCs, fibroblasts, immune cells, endothelial cells), soluble factors (e.g., growth factors, hormones and cytokines), and extracellular matrix. ATM—ataxia–telangiectasia mutated; ATR—ATM- and Rad3-Related; Chk1—checkpoint kinase 1; Chk2—checkpoint kinase 2; ECM—extracellular matrix; FAK—focal adhesion kinase; GSH—glutathione; JAK—Janus kinase; EGFR—epidermal growth factor receptor; HIF—hypoxia-inducible factor; PI3K—phosphatidylinositol 3-kinases; ROS—reactive oxygen species.
To repair DSBs, cells employ two major mechanisms: the more error-prone non-homologous end joining (NHEJ) and the more accurate homologous recombination (HR). In addition, tumor cells also have two extremely error-prone DSB back-up repair mechanisms for both NHEJ and HR, the alt-EJ and single-strand annealing (SSA)
[34]. In mammalian cells, HR occurs only in the late synthesis (S) phase and less in the gap 2 (G2) phase of the cell cycle when the DNA template on the sister chromatid is available for recombination, whereas NHEJ is active throughout the entire cell cycle with highest efficiency during the G2/mitosis (M) stage and is predominant in G0, G1, and early S phases
[35][36][37][38][39].
An activation of these different DNA repair mechanisms at specific phases of the cell cycle results in differences in radiosensitivity throughout the cell cycle, with increased radioresistance in the late S phase and increased radioresponsiveness in G2 and M phases
[40]. Increased cell radioresistance in the S phase was attributed to an increased level of DNA replication enabling the HR process
[40]. Resistance caused by HR-mediated repair is further enhanced by the presence of all available DNA repair pathways, including those that go beyond the repair of DSBs
[39][41]. Therefore, the second “R” refers to redistribution of tumor cells in the cell cycle during radiotherapy to the more radiosensitive cell states. Similar to normal stem cells, some CSC populations are slow-proliferating or quiescent cells
[42]. A comparative analysis of DNA repair in the proliferating and quiescent tissues showed that quiescent cells lack DNA repair efficiency and display sustained accumulation of DSBs that could cause an activation of p53 signaling and apoptosis
[43][44]. In contrast to the normal tissues and tumor bulk, CSCs have attenuated activation of p53 after radiation-induced DNA damage and, as result, impaired cell cycle arrest and apoptosis that, in the long run, might lead to accumulation of DNA mutations
[15][44]. This accumulation of mutation burden over time increases intratumoral heterogeneity and leads to tumor evolution and disease progression
[15]. The CSC evolution during tumor development and treatment is associated with activation of different pro-survival pathways which cause intrinsic radioresistance of tumor cells such as epidermal growth factor receptor (EGFR), phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR), wingless-type MMTV integration site family (WNT), Notch, and Hedgehog signaling
[11][14][45], and different agents targeting these molecular pathways are currently in preclinical and clinical development.
In addition to the intrinsic mechanisms of radioresistance, the fate of tumor cells after radiotherapy depends on the plethora of cues coming from the tumor microenvironment. Tumors show growth comparable to that of rapidly proliferating tissues with a comparably high demand for oxygen and nutrients from the blood vessels. A number of recent studies demonstrated that tumors can make their own blood vessels and some CSC populations, e.g., in glioblastoma, are capable of differentiating into endothelial cells
[46][47]. However, sometimes tumors outgrow their vasculature and develop regions with an inadequate nourishment and supply of oxygen. The hypoxic microenvironment might promote radiotherapy resistance by decreasing DSB burden mediated by oxygen-dependent free-radical production as discussed elsewhere
[48][49]. Hypoxia induces significant changes in tumor metabolism due to the deficiency of nutrients, low concentration of oxygen, and deregulation of transporter proteins and metabolic enzymes
[49]. An impaired mitochondrial respiration in tumor cells under hypoxia increases glucose uptake to cover high energetic demands of growing tumors. This biological feature is used in clinical practice for tumor imaging by positron emission tomography (PET) using radiolabeled glucose analog fluorine-18-2-fluoro-2-deoxy-
d-glucose (
18F-FDG)
[50][51]. Sustained hypoxia can also trigger mechanisms important for the maintenance of CSCs such as hypoxia-inducible factor (HIF) signaling, autophagy, and epithelial–mesenchymal transition (EMT) which is associated with acquisition of the mesenchymal characteristics by tumor cells. Recent data suggest that HIF signaling is important for the regulation of CSC properties in glioblastoma, ovarian cancer, and breast cancer, and also contributes to the activation of autophagy and EMT
[49][52][53][54][55]. Autophagy is a mechanism of cellular “self-eating” by which cells sequestrate and digest certain organelles and protein complexes in a process of clean-up of damaged structures and misfolded proteins
[56]. Autophagy might decrease efficiency of radiotherapy by its contribution to CSC maintenance and reduction of ROS-associated DNA damage
[56][57][58]. A growing body of pre-clinical data demonstrated that EMT-related signaling pathways contribute to tumor cell reprogramming into CSCs, metastatic tumor spread, and radioresistance
[59][60][61][62]. Taken together, these findings suggest that increasing the content of molecular oxygen in the tumor, or tumor reoxygenation, might be an effective way to increase radiation-induced DNA damage and to inhibit the signaling pathways and mechanisms favorable for CSC survival.
Tumor recurrences arise after radiotherapy depending on the actively proliferating tumor progenitor cells induced by tumor reoxygenation
[63]. Tumor cell repopulation in the course of treatment might cause therapy failure. In support of this, clinical studies demonstrated that shortening the overall treatment time for fast-proliferating tumors prevents tumor repopulation and contributes to improved local tumor control
[64]. The unlimited self-renewal capacity of CSCs suggests their role in tumor repopulation between the radiotherapy fractions. The recent computational modeling of HNSCC growth demonstrated that tumor re-oxygenation during treatment can result in substantial increase in the probability of CSC symmetric division (up to 50% as compared to 2% before treatment) that yields an increase of the CSC population within the tumor up to 30–35%
[65].
Hereby, the radiobiological concept of 5Rs has to be considered in context of the cancer stem cell tumor model to explain the effect of radiotherapy on tumor tissues. In the next chapters, we discuss the DNA repair and further mechanisms of CSC therapy resistance to identify them as potential targets for the specific elimination of CSC populations.
3. Molecular Mechanisms of CSC Radioresistance
3.1. DNA Repair Factors and Pathways Upregulated in CSCs
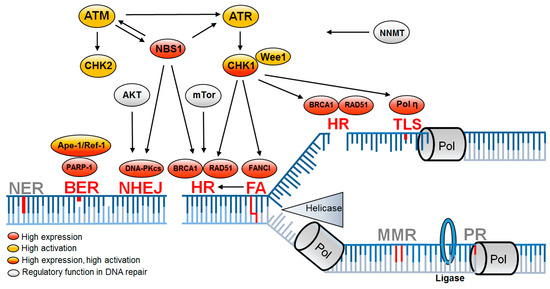
It was previously observed that CSCs, comparable to tissue stem cells, have altered DNA damage response and repair pathways (
Figure 2). In tissue stem cells, the alteration of DNA repair pathways ensures error-free DNA preservation. In CSCs, on the other hand, this causes resistance to exogenously induced DNA damage that leads to the failure of tumor therapy
[66][67]. After irradiation, resistance correlates with significantly less DNA damage in glioma and breast cancer stem cells
[68][69][70]. This also confirms the generally accepted view that tissue stem cells guarantee the maintenance of genomic stability and CSCs ensure the survival of the entire tumor population.
Figure 2. DNA repair pathways altered in CSCs are associated with DNA replication. Replication-associated DNA repair pathways leading to enhanced DNA repair (red) mediate resistance in CSCs, while further replication-associated DNA repair pathways remain unchanged (gray). Upregulation is based on increased expression (red) or increased activation (yellow) or both (red/yellow) and is supported by regulatory proteins (gray). BER—base excision repair; FA—Fanconi anemia; HR—homologous recombination; NER—nucleotide excision repair; NHEJ—non-homologous end joining; MMR—mismatch repair; PR—proof reading; TLS—translesion synthesis
[9][68][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85].
Most of the investigations regarding radioresistance and DNA repair were performed in glioblastoma. Radioresistance is mediated by enhanced activation of the two serine threonine protein kinases ataxia–telangiectasia mutated (ATM) and ATM- and Rad3-Related (ATR) and their two downstream checkpoint kinases (Chk1 and Chk2)
[68][69][82][86][87]. Both kinases regulate the complex DNA damage response by initiating cell-cycle checkpoint control and activating corresponding DNA repair pathways. CD133
+ glioma stem cells show an increased Chk1-dependent checkpoint response
[9][69][86]. An increased expression of
NBS1, a component of the Mre11, Rad50, Nbs1 (MRN) complex
[88] involved in DSB sensor activity, was also detected. Increased DNA damage response and repair gene expression were also observed in CD133
+ CSCs in lung carcinoma cells, breast, breast carcinoma-inducing cells, pancreatic CSCs, and non-small-cell lung cancer
[41][71].
Starting point for the enhanced DNA damage response appears to be a general adaptation to increased replication stress and increased oxidative damage already in the untreated state, which leads to increased activation of the DNA damage response even after irradiation
[84][89]. The DNA damage response is then mediated either by induced DSBs for amplified activation of ATM or by increased replicative stress for amplified activation of ATR. This results in a lower number of DSBs
[89][90].
With regard to DNA double-strand break repair pathways in CSCs, HR is of outstanding importance, whereas, for NHEJ, only ATM-mediated effects are observed
[91][92][93]. HR is the preferred DSB repair pathway in the S phase and it is more strongly activated by ATR and ATM in cells tolerating replication stress
[68][72]. RAD51 overexpression, the main HR DNA repair protein, is observed in glioma stem cells and decreases at the transition to progenitor cells
[92]. Glioblastoma cell lines show an upregulation of HR genes, especially
RAD51,
BRCA1, and
BRCA2, which leads to less DNA damage after irradiation
[74]. Thus, most studies suggest that resistance of CSCs manifests during the S phase by HR, regulated by MRN and activated by ATM/ATR.
The importance of S-phase repair also appears for CSC resistance mechanisms after chemotherapy in ovarian cancer. Here, resistance is observed by enhanced activation of translesion synthesis mediated by polymerase eta
[79], as well as in HNSCC, where overexpression of the Fanconi anemia DNA repair proteins was observed in ALDH1-positive tumor cells
[81].
However, a number of studies showed no difference or even lower DNA damage response in CSCs
[41][91][94][95][96]. These contradictory observations suggest that an improved DNA damage response may not be a common feature of CSCs. More obvious is that CSCs and non-CSCs are transient populations and that, in addition to inter-tumoral heterogeneity, intratumoral heterogeneity must also be considered in DNA damage reaction functionality
[96]. This was previously observed in glioblastoma, where
RAD51 expression decreases from CSC to progenitor cells
[92].
3.2. Factors Indirectly Influencing DNA Repair Capacity of CSCs
In CSCs, radioresistance determined by DNA repair capacity may also be attributed to lower indirectly induced ROS causing DNA damage and ROS-dependent apoptosis
[14][70]. Breast CSCs express higher concentrations of ROS scavengers and neutralize radiation-induced ROS
[89]. In addition to the known proteins with ROS scavenger function, the multifunctional protein apurine/apirimidine endonuclease/redox effector factor (Ape1/Ref-1) is also increasingly expressed in CSCs. Among other functions, Ape1/Ref-1 is part of the DNA repair complex base excision repair (BER), so that Ape1/Ref-1 can reduce both intracellular ROS and increase DNA repair
[68]. Radioresistance in mesenchymal CSCs indirectly influencing DNA repair capacity could also be due to nicotinamide
N-methyltransferase (NNMT) overexpression through depletion of the accessible amounts of nicotinamide, which is a known inhibitor of cellular DNA repair mechanisms
[85].
An additional resistance mechanism only indirectly linked to improved DNA repair is the induction of CSC quiescence. By stopping the cells in the G0 phase, DNA damage can be eliminated before entering the S phase
[44][97]. This shift in the cell cycle distribution may explain why NHEJ was considered important for increased radiation resistance in recent studies
[68][76][98][99][100].
Another observed phenomenon indirectly linked to DNA repair mediated resistance is that genotoxic treatment, such as irradiation and chemotherapy, itself leads to the reprogramming of non-CSCs into CSCs
[22][75][83][101][102][103][104][105]. In reprogrammed stem-like cells, either a faster repair mediated by
BRCA1 and
RAD51 after gemcitabine in pancreatic cancer
[75] or a stronger activation of ATR/Chk1 in colon carcinoma after treatment with DNA interstrand-crosslinking (ICL) agents was shown
[83]. Zhang and colleagues even went so far as to postulate a direct dependence of the DNA signaling cascade and stem-cell characteristics. They observed an ATM-mediated stabilization of zinc finger E-box binding homeobox 1 (ZEB1) leading to an enhanced Chk1-dependent DNA damage response in previously epithelial breast cells
[104]. This direct dependence on stem cell character and HR or S-phase DNA repair was also observed for breast epithelial cells. Depletion of
BRCA1 and
FANCD2 led to reprogramming in breast epithelial cells to mesenchymal phenotype
[105].
 +1 point
+1 point