 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Amanda M Levy | + 2877 word(s) | 2877 | 2021-09-07 04:12:53 | | | |
| 2 | Lindsay Dong | Meta information modification | 2877 | 2021-09-08 03:11:52 | | | | |
| 3 | Lindsay Dong | Meta information modification | 2877 | 2021-09-15 09:47:08 | | |
Video Upload Options
Gilles de la Tourette syndrome (GTS) is a childhood-onset neurodevelopmental and -psychiatric tic-disorder of complex etiology which is often comorbid with obsessive-compulsive disorder (OCD) and/or attention deficit hyperactivity disorder (ADHD). Twin and family studies of GTS individuals have shown a high level of heritability suggesting, that genetic risk factors play an important role in disease etiology. However, the identification of major GTS susceptibility genes has been challenging, presumably due to the complex interplay between several genetic factors and environmental influences, low penetrance of each individual factor, genetic diversity in populations, and the presence of comorbid disorders. Even though several strong candidate genes have hitherto been identified, none of these have turned out to be major susceptibility genes yet.
1. Introduction
2. Candidate Gene and Pathway
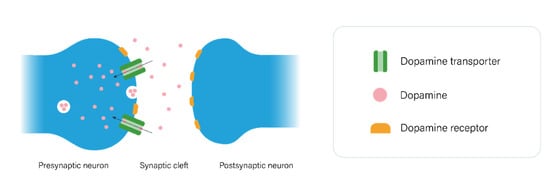
2.1. The Dopaminergic Pathway
2.2. The Serotonergic Pathway
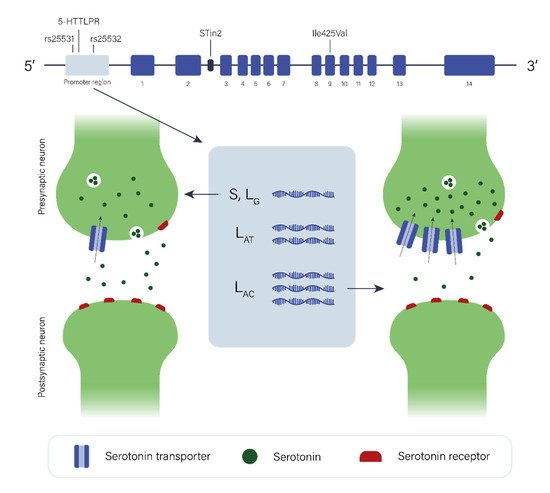
The data regarding the role of the serotonergic pathway in GTS etiology are limited and again, individual studies have been of limited statistical power with low sample size. However, when studies of the dopaminergic system are taken into consideration, it is plausible to hypothesize that the serotonergic system may play a complex role in GTS etiology, directly and/or indirectly in combination with or through modulating dopaminergic neurotransmission. Additional neurotransmitter pathways (e.g., glutamatergic and GABAergic) may also be involved in the pathology of GTS, as the dysfunction of one pathway may affect others due to interaction or self-regulation among them.
3. Linkage Analyses, GWAS & Other Studies
3.1. SLITRK1
SLITRK1 promotes neurite outgrowth and SLITRK1 is predominantly expressed in the brain, including the CBGTC circuits [34]. In addition, the six proteins belonging to the SLITRK family play important roles in the development of the central nervous system and neuronal processes and have been implicated in several other neurodevelopmental and -psychiatric disorders [35]. The SLITRK-genes have therefore been attractive candidates in the search for the genes involved in GTS etiology. However, it is likely that these genes, especially SLITRK1, do not play a major role in GTS etiology.
3.2. IMMPL2
3.3. HDC
3.4. CELSR3, WWC1, FN1, and NIPBL
3.5. ASH1L
3.6. FLT3
3.7. NRXN1 and CNTN6
4. Conclusions
References
- Knight, T.; Steeves, T.; Day, L.; Lowerison, M.; Jette, N.; Pringsheim, T. Prevalence of Tic Disorders: A Systematic Review and Meta-Analysis. Pediatr. Neurol. 2012, 47, 77–90.
- Scharf, J.M.; Miller, L.L.; Bs, C.A.G.; Ma, J.A.; Mathews, C.A.; Ben-Shlomo, Y. Population prevalence of Tourette syndrome: A systematic review and meta-analysis. Mov. Disord. 2014, 30, 221–228.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013.
- Müller-Vahl, K.R.; Sambrani, T.; Jakubovski, E. Tic disorders revisited: Introduction of the term “tic spectrum disorders”. Eur. Child Adolesc. Psychiatry 2019, 28, 1129–1135.
- Conte, G.; Valente, F.; Fioriello, F.; Cardona, F. Rage attacks in Tourette Syndrome and Chronic Tic Disorder: A systematic review. Neurosci. Biobehav. Rev. 2020, 119, 21–36.
- Darrow, S.M.; Grados, M.; Sandor, P.; Hirschtritt, M.E.; Illmann, C.; Osiecki, L.; Dion, Y.; King, R.; Pauls, D.; Budman, C.L.; et al. Autism Spectrum Symptoms in a Tourette’s Disorder Sample. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 610–617.e1.
- Huisman-van Dijk, H.M.; van de Schoot, R.; Rijkeboer, M.M.; Mathews, C.A.; Cath, D.C. The relationship between tics, OC, ADHD and autism symptoms: A cross- disorder symptom analysis in Gilles de la Tourette syndrome patients and family-members. Psychiatry Res. 2016, 237, 138–146.
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agundez, J. Sleep disorders in tourette syndrome. Sleep Med. Rev. 2020, 53, 101335.
- Robertson, M.M.; Eapen, V.; Singer, H.S.; Martino, D.; Scharf, J.M.; Paschou, P.; Roessner, V.; Woods, D.W.; Hariz, M.; Mathews, C.A.; et al. Gilles de la Tourette syndrome. Nat. Rev. Dis. Prim. 2017, 3, 16097.
- Vermilion, J.; Pedraza, C.; Augustine, E.F.; Adams, H.R.; Vierhile, A.; Lewin, A.B.; Collins, A.T.; McDermott, M.P.; O’Connor, T.; Kurlan, R.; et al. Anxiety Symptoms Differ in Youth With and Without Tic Disorders. Child Psychiatry Hum. Dev. 2021, 52, 301–310.
- Hoekstra, P.J.; Dietrich, A.; Edwards, M.J.; Elamin, I.; Martino, D. Environmental factors in Tourette syndrome. Neurosci. Biobehav. Rev. 2013, 37, 1040–1049.
- Frick, L.; Pittenger, C. Microglial Dysregulation in OCD, Tourette Syndrome, and PANDAS. J. Immunol. Res. 2016, 2016, 1–8.
- Paschou, P. The genetic basis of Gilles de la Tourette Syndrome. Neurosci. Biobehav. Rev. 2013, 37, 1026–1039.
- Draganski, B.; Martino, D.; Cavanna, A.E.; Hutton, C.; Orth, M.; Robertson, M.M.; Critchley, H.D.; Frackowiak, R.S. Multispectral brain morphometry in Tourette syndrome persisting into adulthood. Brain 2010, 133, 3661–3675.
- Peterson, B.S.; Thomas, P.; Kane, M.J.; Scahill, L.; Zhang, H.; Bronen, R.; King, R.A.; Leckman, J.F.; Staib, L. Basal Ganglia Volumes in Patients With Gilles de la Tourette Syndrome. Arch. Gen. Psychiatry 2003, 60, 415–424.
- Wang, Z.; Maia, T.; Marsh, R.; Colibazzi, T.; Gerber, A.; Peterson, B.S. The Neural Circuits That Generate Tics in Tourette’s Syndrome. Am. J. Psychiatry 2011, 168, 1326–1337.
- Neuner, I.; Schneider, F.; Shah, N.J. Functional Neuroanatomy of Tics. In International Review of Neurobiology; Harris, R.A., Jenner, P., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 35–71.
- Cumming, P. The life history of dopamine. In Imaging Dopamine; Cambridge University Press: Cambridge, UK, 2009; pp. 5–18.
- Beaulieu, J.-M.; Gainetdinov, R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217.
- Sinopoli, V.M.; Burton, C.; Kronenberg, S.; Arnold, P.D. A review of the role of serotonin system genes in obsessive-compulsive disorder. Neurosci. Biobehav. Rev. 2017, 80, 372–381.
- Olivier, B. Serotonin: A never-ending story. Eur. J. Pharmacol. 2015, 753, 2–18.
- Bortolozzi, A.; Diaz-Mataix, L.; Scorza, M.C.; Celada, P.; Artigas, F. The activation of 5-HT2A receptors in prefrontal cortex enhances dopaminergic activity. J. Neurochem. 2005, 95, 1597–1607.
- De Deurwaerdère, P.; Navailles, S.; Berg, K.A.; Clarke, W.P.; Spampinato, U. Constitutive Activity of the Serotonin2C Receptor Inhibits In Vivo Dopamine Release in the Rat Striatum and Nucleus Accumbens. J. Neurosci. 2004, 24, 3235–3241.
- Esposito, E.; Di Matteo, V.; Di Giovanni, G. Serotonin–dopamine interaction: An overview. Prog. Brain Res. 2008, 172, 3–6.
- Larsen, M.B.; Sonders, M.S.; Mortensen, O.V.; Larson, G.A.; Zahniser, N.R.; Amara, S.G. Dopamine Transport by the Serotonin Transporter: A Mechanistically Distinct Mode of Substrate Translocation. J. Neurosci. 2011, 31, 6605–6615.
- Moya, P.; Wendland, J.R.; Rubenstein, L.M.; Timpano, K.; Heiman, G.; Tischfield, J.; King, R.A.; Andrews, A.; Ramamoorthy, S.; McMahon, F.; et al. Common and rare alleles of the serotonin transporter gene, SLC6A4, associated with Tourette’s disorder. Mov. Disord. 2013, 28, 1263–1270.
- Hildonen, M.; Levy, A.M.; Dahl, C.; Bjerregaard, V.A.; Møller, L.B.; Guldberg, P.; Debes, N.M.; Tümer, Z. Elevated Expression of SLC6A4 Encoding the Serotonin Transporter (SERT) in Gilles de la Tourette Syndrome. Genes 2021, 12, 86.
- Dehning, S.; Müller, N.; Matz, J.; Bender, A.; Kerle, I.; Benninghoff, J.; Musil, R.; Spellmann, I.; Bondy, B.; Möller, H.-J.; et al. A genetic variant of HTR2C may play a role in the manifestation of Tourette syndrome. Psychiatr. Genet. 2010, 20, 35–38.
- Cavallini, M.C.; Di Bella, D.; Catalano, M.; Bellodi, L. An association study between 5-HTTLPR polymorphism, COMT polymorphism, and Tourette’s syndrome. Psychiatry Res. 2000, 97, 93–100.
- Liu, S.; Zhang, X.; Yin, Y.; Wang, M.; Che, F.; Ma, X. An Association Analysis between 5-HTTLPR Polymorphism and Obsessive-Compulsive Disorder, Tourette Syndrome in a Chinese Han Population. CNS Neurosci. Ther. 2011, 17, 793–795.
- Hu, X.-Z.; Lipsky, R.; Zhu, G.; Akhtar, L.A.; Taubman, J.; Greenberg, B.D.; Xu, K.; Arnold, P.D.; Richter, M.A.; Kennedy, J.L.; et al. Serotonin Transporter Promoter Gain-of-Function Genotypes Are Linked to Obsessive-Compulsive Disorder. Am. J. Hum. Genet. 2006, 78, 815–826.
- Cao, X.; Zhang, Y.; Abdulkadir, M.; Deng, L.; Fernandez, T.V.; Julie, B.G.; Pieter, H.; Robert, J.H.; Justin, A.K.; Kuperman, S.; et al. Whole-exome sequencing identifies genes associated with Tourette’s disorder in multiplex families. Mol. Psychiatry 2021, 1–15.
- Abelson, J.F.; Kwan, K.Y.; O’Roak, B.J.; Baek, D.Y.; Stillman, A.A.; Morgan, T.M.; Mathews, C.A.; Pauls, D.L.; Rašin, M.-R.; Gunel, M.; et al. Sequence Variants in SLITRK1 Are Associated with Tourette’s Syndrome. Science 2005, 310, 317–320.
- Stillman, A.A.; Krsnik, Ž.; Sun, J.; Rašin, M.-R.; State, M.W.; Şestan, N.; Louvi, A. Developmentally regulated and evolutionarily conserved expression of SLITRK1 in brain circuits implicated in Tourette syndrome. J. Comp. Neurol. 2009, 513, 21–37.
- Proenca, C.C.; Gao, K.P.; Shmelkov, S.V.; Rafii, S.; Lee, F.S. Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends Neurosci. 2011, 34, 143–153.
- Petek, E.; Windpassinger, C.; Vincent, J.B.; Cheung, J.; Boright, A.P.; Scherer, S.; Kroisel, P.M.; Wagner, K. Disruption of a Novel Gene (IMMP2L) by a Breakpoint in 7q31 Associated with Tourette Syndrome. Am. J. Hum. Genet. 2001, 68, 848–858.
- Boghosian-Sell, L.; Comings, D.E.; Overhauser, J. Tourette syndrome in a pedigree with a 7;18 translocation: Identification of a YAC spanning the translocation breakpoint at 18q22.3. Am. J. Hum. Genet. 1996, 59, 999–1005.
- Patel, C.J.; Cooper-Charles, L.; McMullan, D.J.; Walker, J.M.; Davison, V.; Morton, J.E. Translocation breakpoint at 7q31 associated with tics: Further evidence for IMMP2L as a candidate gene for Tourette syndrome. Eur. J. Hum. Genet. 2011, 19, 634–639.
- Díaz-Anzaldúa, A.; Joober, R.; Rivière, J.-B.; Dion, Y.; Lespérance, P.; Chouinard, S.; Richer, F.; Rouleau, G.A. Association between 7q31 markers and tourette syndrome. Am. J. Med. Genet. Part A 2004, 127A, 17–20.
- Pagliaroli, L.; Vereczkei, A.; Padmanabhuni, S.S.; Tarnok, Z.; Farkas, L.; Nagy, P.; Rizzo, R.; Wolanczyk, T.; Szymanska, U.; Kapisyzi, M.; et al. Association of Genetic Variation in the 3′UTR of LHX6, IMMP2L, and AADAC With Tourette Syndrome. Front. Neurol. 2020, 11, 803.
- Haas, H.L.; Sergeeva, O.A.; Selbach, O. Histamine in the Nervous System. Physiol. Rev. 2008, 88, 1183–1241.
- Ercan-Sencicek, A.G.; Stillman, A.A.; Ghosh, A.K.; Bilguvar, K.; O’Roak, B.J.; Mason, C.E.; Abbott, T.; Gupta, A.; King, R.A.; Pauls, D.L.; et al. L-Histidine Decarboxylase and Tourette’s Syndrome. N. Engl. J. Med. 2010, 362, 1901–1908.
- Alexander, J.; Potamianou, H.; Xing, J.; Deng, L.; Karagiannidis, I.; Tsetsos, F.; Drineas, P.; Tarnok, Z.; Rizzo, R.; Wolanczyk, T.; et al. Targeted Re-Sequencing Approach of Candidate Genes Implicates Rare Potentially Functional Variants in Tourette Syndrome Etiology. Front. Neurosci. 2016, 10, 428.
- Depienne, C.; Ciura, S.; Trouillard, O.; Bouteiller, D.; Leitão, E.; Nava, C.; Keren, B.; Marie, Y.; Guegan, J.; Forlani, S.; et al. Association of Rare Genetic Variants in Opioid Receptors with Tourette Syndrome. Tremor Other Hyperkinet. Mov. (N. Y.) 2019, 9.
- Karagiannidis, I.; Dehning, S.; Sandor, P.; Tarnok, Z.; Rizzo, R.; Wolanczyk, T.; Madruga-Garrido, M.; Hebebrand, J.; Nöthen, M.; Lehmkuhl, G.; et al. Support of the histaminergic hypothesis in Tourette Syndrome: Association of the histamine decarboxylase gene in a large sample of families. J. Med. Genet. 2013, 50, 760–764.
- Baldan, L.C.; Williams, K.A.; Gallezot, J.-D.; Pogorelov, V.; Rapanelli, M.; Crowley, M.; Anderson, G.M.; Loring, E.; Gorczyca, R.; Billingslea, E.; et al. Histidine Decarboxylase Deficiency Causes Tourette Syndrome: Parallel Findings in Humans and Mice. Neuron 2014, 81, 77–90.
- Fernandez, T.V.; Sanders, S.; Yurkiewicz, I.R.; Ercan-Sencicek, A.G.; Kim, Y.-S.; Fishman, D.O.; Raubeson, M.J.; Song, Y.; Yasuno, K.; Ho, W.S.; et al. Rare Copy Number Variants in Tourette Syndrome Disrupt Genes in Histaminergic Pathways and Overlap with Autism. Biol. Psychiatry 2012, 71, 392–402.
- Lei, J.; Deng, X.; Zhang, J.; Su, L.; Xu, H.; Liang, H.; Huang, X.; Song, Z.; Deng, H. Mutation screening of the HDC gene in Chinese Han patients with Tourette syndrome. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 159B, 72–76.
- Dong, H.; Liu, W.; Liu, M.; Xu, L.; Li, Q.; Zhang, R.; Zhang, X.; Liu, S. Investigation of a Possible Role for the Histidine Decarboxylase Gene in Tourette Syndrome in the Chinese Han Population: A Family-Based Study. PLoS ONE 2016, 11, e0160265.
- Willsey, A.J.; Fernandez, T.V.; Yu, D.; King, R.A.; Dietrich, A.; Xing, J.; Sanders, S.J.; Mandell, J.D.; Huang, A.Y.; Richer, P.; et al. De Novo Coding Variants Are Strongly Associated with Tourette Disorder. Neuron 2017, 94, 486–499.e9.
- Wang, S.; Mandell, J.D.; Kumar, Y.; Sun, N.; Morris, M.T.; Arbelaez, J.; Nasello, C.; Dong, S.; Duhn, C.; Zhao, X.; et al. De Novo Sequence and Copy Number Variants Are Strongly Associated with Tourette Disorder and Implicate Cell Polarity in Pathogenesis. Cell Rep. 2018, 24, 3441–3454.e12.
- gnomAD. Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org/ (accessed on 19 February 2021).
- Liu, S.; Tian, M.; He, F.; Li, J.; Xie, H.; Liu, W.; Zhang, Y.; Zhang, R.; Yi, M.; Che, F.; et al. Mutations in ASH1L confer susceptibility to Tourette syndrome. Mol. Psychiatry 2020, 25, 476–490.
- Yu, D.; Sul, J.H.; Tsetsos, F.; Nawaz, M.S.; Huang, A.Y.; Zelaya, I.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; et al. Interrogating the Genetic Determinants of Tourette’s Syndrome and Other Tic Disorders Through Genome-Wide Association Studies. Am. J. Psychiatry 2019, 176, 217–227.
- Tsetsos, F.; Yu, D.; Sul, J.H.; Huang, A.Y.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; Greenberg, E.; Muller-Vahl, K.R.; et al. Synaptic processes and immune-related pathways implicated in Tourette syndrome. Transl. Psychiatry 2021, 11, 1–12.
- Malhotra, D.; Sebat, J. CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. Cell 2012, 148, 1223–1241.
- Huang, A.Y.; Yu, D.; Davis, L.K.; Sul, J.H.; Tsetsos, F.; Ramensky, V.; Zelaya, I.; Ramos, E.M.; Osiecki, L.; Chen, J.A.; et al. Rare Copy Number Variants in NRXN1 and CNTN6 Increase Risk for Tourette Syndrome. Neuron 2017, 94, 1101–1111.e7.
- Mercati, O.; Huguet, G.; Danckaert, A.; André-Leroux, G.; Maruani, A.; Bellinzoni, M.; Rolland, T.; Gouder, L.; Mathieu, A.; Buratti, J.; et al. CNTN6 mutations are risk factors for abnormal auditory sensory perception in autism spectrum disorders. Mol. Psychiatry 2017, 22, 625–633.
- Kashevarova, A.A.; Nazarenko, L.P.; Schultz-Pedersen, S.; Skryabin, N.A.; Salyukova, O.A.; Chechetkina, N.N.; Tolmacheva, E.N.; Rudko, A.A.; Magini, P.; Graziano, C.; et al. Single gene microdeletions and microduplication of 3p26.3 in three unrelated families: CNTN6 as a new candidate gene for intellectual disability. Mol. Cytogenet. 2014, 7, 97.
- Pak, C.; Danko, T.; Zhang, Y.; Aoto, J.; Anderson, G.; Maxeiner, S.; Yi, F.; Wernig, M.; Südhof, T.C. Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell 2015, 17, 316–328.
- Sundaram, S.K.; Huq, A.M.; Wilson, B.J.; Chugani, H.T. Tourette syndrome is associated with recurrent exonic copy number variants. Neurology 2010, 74, 1583–1590.
- Nag, A.; Bochukova, E.; Kremeyer, B.; Campbell, D.; Muller, H.; Valencia-Duarte, A.V.; Cardona, J.; Rivas, I.C.; Mesa, S.C.; Cuartas, M.; et al. CNV Analysis in Tourette Syndrome Implicates Large Genomic Rearrangements in COL8A1 and NRXN1. PLoS ONE 2013, 8, e59061.


