Due to the consequences of genome instability in cancer cells, sometimes two mRNAs can be fused to generate chimeric RNAs. Several recent studies demonstrated that chimeric RNAs are significantly associated with oncogenesis and can also promote drug resistance. The generation of chimeric RNAs could allow cancer cells to switch their functionality. Therefore, chimeric RNAs are an important driver for generating the phenotypic plasticity of cancer cells and increasing their fitness in the tissue environment. Chimeric RNAs could be translated and generate new fusion or chimeric proteins that could alter the normal pathways and lead to cancer development. Chimeric RNAs could also generate long non-coding RNA (lncRNA), which could regulate cancer cell proliferation.
1. Overview
Fusion of exons or introns from two different genes can lead to the formation of chimeric RNAs. Several recent studies have reported that chimeric RNAs promote tumorigenesis and cancer drug resistance. Therefore, chimeric RNAs are crucial for generating phenotypic diversity between cancer cells that drives the adaptive evolution of cancer.
Gene fusions can give rise to somatic alterations in cancers. Fusion genes have the potential to create chimeric RNAs, which can generate the phenotypic diversity of cancer cells, and could be associated with novel molecular functions related to cancer cell survival and proliferation. The expression of chimeric RNAs in cancer cells might impact diverse cancer-related functions, including loss of apoptosis and cancer cell plasticity, and promote oncogenesis. Due to their recurrence in cancers and functional association with oncogenic processes, chimeric RNAs are considered biomarkers for cancer diagnosis. Several recent studies demonstrated that chimeric RNAs could lead to the generation of new functionality for the resistance of cancer cells against drug therapy. Therefore, targeting chimeric RNAs in drug resistance cancer could be useful for developing precision medicine. So, understanding the functional impact of chimeric RNAs in cancer cells from an evolutionary perspective will be helpful to elucidate cancer evolution, which could provide a new insight to design more effective therapies for cancer patients in a personalized manner.
2. Cancer Evolution
Traditionally, cancer development has been accepted as a multistage process driven by the stepwise accumulation of new genetic changes, which promotes the gaining of several abilities that allow cancer cells to survive and proliferate without being subjected to cellular regulatory barriers
[1][2][3]. In recent years, however, it has become clear that stochastic cellular macroevolution appears suddenly by saltation for most cancer types, which challenges the neo-Darwinian concept of cancer evolution. Cancer formation requires macroevolution, as only new systems can break a series of barriers from normal tissues/organs/body, where various constraints, including cellular, tissue, and immune factors, prevent the phase transition from a normal cell to cancer
[4]. Cancer cells evolve all the way through disease progression, metastasis, and tumor relapse via multiple cycles of two-phased cancer evolution (genome alteration-mediated macroevolution, followed by gene mutation-mediated microevolution)
[5][6][7][8]. Cancer evolution is a dynamic process involving genotypic and phenotypic changes, ensuring the high level of plasticity of cancer cells
[8][9][10]. Such plasticity, or heterogeneity, is the lifeline for cancer cells, as it helps cancer cells to survive and become dominant under multiple levels of constraints. Constant change is the winning strategy for cancer cells, and genome instability is a powerful mechanism that allows both the survival (by changing genome structure within the macroevolutionary phase) and fitness (by changing gene mutation/epigenetic profile within the microevolutionary phase) of cancer cells
[11][12][13][14]. Genomic instability can give rise to gene mutations, chromosomal translocations, alternations of copy number, deletions, and inversions of pieces of DNA
[15]. Genomic instability is an important mechanism that enables the acquisition of new characteristics required for oncogenesis, which is the potential driver of cancer evolution
[16].
Due to the consequences of genome instability in cancer cells, sometimes two mRNAs can be fused to generate chimeric RNAs
[17]. Several recent studies demonstrated that chimeric RNAs are significantly associated with oncogenesis
[18][19] and can also promote drug resistance
[20][21][22]. The generation of chimeric RNAs could allow cancer cells to switch their functionality. Therefore, chimeric RNAs are an important driver for generating the phenotypic plasticity of cancer cells and increasing their fitness in the tissue environment. Chimeric RNAs could be translated and generate new fusion or chimeric proteins that could alter the normal pathways and lead to cancer development
[19][23][24]. Chimeric RNAs could also generate long non-coding RNA (lncRNA), which could regulate cancer cell proliferation
[18][25][26].
3. Mechanisms of Formation of Chimeric RNAs in Cancer Cells and Their Functional Associations with Cancer Development
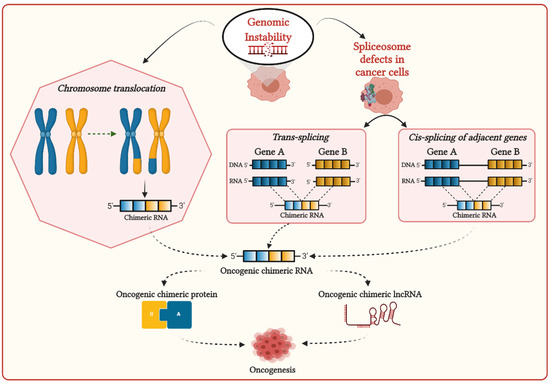
Genomic instability can induce chromosomal aberrations such as translocation, enabling the generation of fusion genes, then transcribe them to corresponding chimeric RNAs (
Figure 1)
[27]. Most chimeric RNAs generated by chromosomal translocation are recurrent and translated in chimeric proteins, which are significantly associated with cancer development
[28]. Chimeric RNAs generated by chromosomal aberrations are prevalently observed in hematopoietic malignancies and sarcomas, frequently involving genes required for chromatin regulation and transcriptional control. Chimeric proteins generated by these chimeric RNAs are thought to be the principal driver of oncogenesis, altering chromatin dynamics to activate the oncogene expressions
[29].
Figure 1. Possible mechanisms for the generation of chimeric transcripts in cancer cells.
The first reported fusion gene BCR-ABL was discovered in human chronic myelogenous leukemia (CML)
[30], which is generated by translocation between the q arms of chromosomes 9 and 22 and is denoted as t(9;22). Fusion protein produced from this BCR-ABL chimeric transcript altered constitutively active ABL1 kinase that can promote the development of CML
[31]. Another chromosomal translocation t(15;17) was detected in acute promyelocytic leukemia (APL), resulting in the formation of promyelocytic leukemia–retinoic acid receptor α (PML-RARα) fusion oncoprotein that can interplay with retinoic X receptors (RXR) and promote the deregulation of epigenetic modifications
[32][33][34]. A recurrent gene fusion TMPRSS2-ERG was observed in more than fifty percent of prostate cancer cases with the deletion of del(q22) and t(7;21)(1,26–28), resulting in translocation of the ERG gene (21q22.3) or the ETV1 gene (7p21.2) to the TMPRSS2 gene (21q22.2) promoter region
[35][36]. This fusion leads to the overexpression of the oncogene ERG or ETS transcription factors in response to androgens induced by the TMPRSS2 promoter, which promotes the generation of molecular heterogeneity and the formation of high-grade tumors
[34][37][38]. In Burkitt lymphoma, three translocations t(8;14)(q24;q32), t(2;8)(p11;q24), or t(8;22)(q24;q11) were observed, where, in all cases, the breakpoint in chromosome 8 is within the MYC gene, and the other breakpoint is within an immunoglobulin gene
[39][40]. These translocations promote the MYC gene to become continuously expressed due to the impact of regulatory elements of the immunoglobulin genes, which are crucial for initiating oncogenesis
[27]. Several fusions associated with the MLL1 gene were found in acute leukemia, which was generated due to recurrent chromosomal rearrangements involving 11q23
[41][42][43][44]. MLL1 fusion-positive leukemia has a remarkably low somatic mutation rate, suggesting that MLL1 fusions are the potential drivers of cancer development
[44][45][46]. Altogether, it can be suggested that the appearance of chimeric RNAs due to chromosomal translocation promotes phenotypic plasticity for cancer development.
The interplay of splicing factors and RNA-binding proteins (RBPs) are found to play an important role in DNA–RNA hybrid (R-loop) formation during transcription to prevent RNA-induced genome instability
[47]. In cancers, mutations in the spliceosome machinery can affect R-loop formation, promoting genomic instability
[48][49][50]. Genome instability and mutations in spliceosome machinery can stimulate aberrant splicing, including cis- and trans-splicing, leading to the generation of chimeric RNAs in cancer cells (
Figure 1)
[51]. Cis-splicing is the mechanism by which two neighboring genes in the same strands are transcribed in the same orientation and generate read-through chimeric transcripts
[52]. The recurrent read-through chimeric transcripts generated via cis-splicing were prevalently observed in renal carcinoma
[53], prostate cancer
[54], and breast cancer
[55]. In prostate cancer, SLC45A3–ELK4 is the most common chimeric RNA generated by cis-splicing
[56][57]. This SLC45A3-ELK4 acts as lncRNA and regulates the proliferation of prostate cancer cells
[25]. This fusion is recurrent and regarded as a potential biomarker for prostate cancer. Another method for generating chimeric RNAs is trans-splicing, by which two individual pre-mRNA molecules can be fused. Although trans-splicing is considered a noncanonical splicing process in humans, recent studies demonstrated that trans-splicing could be involved in generating chimeric RNAs in human cells. The two most common examples are JAZF1–SUZ12
[58][59] in endometrial stromal tumors and PAX3–FOXO1
[60] in rhabdomyosarcoma, where, in both cases, identical chimeric RNAs were found as chromosomal translocation from cancer cells and RNA trans-splicing from normal human cells. Therefore, the generation of identical chimeric RNAs in cancer cells by chromosomal translocation, which is also generated in normal cells due to different mechanisms, could potentially be associated with distinct pathological consequences in cancers.
4. Conclusions
From the beginning of the discovery of chimeric RNAs, they have been found to be associated with cancers. With the advancement of high-throughput sequencing technology and cancer genomics, several chimeric RNAs have been identified in different cancer samples, where some chimeric RNAs were found recurrent and some specific to the sample. As discussed above, several chimeric RNAs were found to be associated with oncogenesis, and several were reported to enable drug resistance in tumors, which supports their clinical significance for targeting them to design new cancer treatments. Further, chimeric proteins generated from these chimeric RNAs have different structures from their parental proteins, which helps them to alter the protein interaction networks in cancer cells that enable their survival and proliferation. Therefore, designing drugs that could target chimeric proteins could be helpful in cancer therapy. Hence, understanding the appearance of chimeric RNAs could guide the direction of cancer evolution, which could be useful to the development of a new strategy for cancer treatment.
As cancer is an evolutionary process, cancer cells should undergo adaptive evolution and create phenotypic diversity to fit into a new environment. The generation of chimeric RNAs is associated with the functional expansion of cancer cells and the creation of phenotypic diversity. Selection directly acts on phenotypes instead of genotypes, and overall phenotypic traits of the cell could determine its perseverance and fate in a cell population. Therefore, the production of chimeric RNAs is important for generating the phenotypic plasticity of cancer cells that can provide a fitness advantage to adapt to new environmental conditions. However, there are several open questions regarding (1) how selection can choose partner genes for chimeric RNAs, (2) how chimeric RNAs can contribute to the fitness of cancer cells, (3) what the mechanism for drug resistance by generating chimeric RNAs is, (4) what the evolutionary principles for some chimeric RNAs are recurrent, and (5) what the fate of chimeric RNAs in cancer cells is. Potential solutions to each of these questions need the understanding of chimeric RNAs through the lens of evolution with the communication of clinical studies. This could help to decipher cancer evolution, which could facilitate the improvement of personalized treatment strategies.
 +1 point
+1 point