1. Introduction
Curcumin (diferuloylmethane; 1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione) is an active constituent derived from the powdered rhizome of Curcuma longa
[1]. This natural product, which belongs to the polyphenols, is the spice (curry, turmeric) commonly used in Asian cuisine and represents a widely studied nutraceutical and the most popular dietary supplement in the world. Curcumin has attracted increasing scientific and clinical interest thanks to its wide range of beneficial functions, including its antioxidant, anti-inflammatory, antiproliferative, antitumor, analgesic, cholesterol-lowering, hemostatic, antidiabetic, and antiamyloid roles. Other beneficial functions include its cyto-, gastro-, and neuro-protective roles as well as antiviral and antibacterial functions
[2][3][4][5]. Curcumin is particularly attractive as a potent therapeutic substance, being a non-mutagenic and non-genotoxic agent, although scarcely bioavailable because of its hydrophobic nature
[6]. In general, in humans, curcumin is recognized as a safe bioactive compound, even if used in high doses
[5]. Several studies suggest that curcumin, like other polyphenols, is a highly pleiotropic molecule that interacts simultaneously with a wide range of molecular targets and influences numerous biochemical and molecular cascades, modulating the activation of various transcription factors and thus regulating the expression of growth factors, receptor complexes, cytokines, and enzymes involving cell proliferation and apoptosis
[1][7][8]. The beneficial effect of curcumin in modulating autophagy through various cell signals, such as PI3K/Akt/mTOR, AMPK, MAPK/ERK1/2, Bcl-2, and Rab GTPase network, has also been demonstrated
[4]. Autophagy is a lysosomal catabolic mechanism critical in maintaining cellular homeostasis under both physiological and pathological conditions
[9] and represents the process by which cells adapt their metabolism to conditions of environmental or intracellular stress
[10]. The multi-step nature of autophagy causes its susceptibility to being damaged at different levels, and its defective activity has been linked to a variety of human diseases
[11][12]. Over the years, increasing evidence has accumulated on the dysfunction of autophagy in neurological and neuromuscular diseases, showing how the dysregulation of autophagy initiation, autophagosome formation, maturation, and the autophagosome-lysosome fusion phase contribute to the pathogenesis of these disorders. Because of its numerous beneficial properties, curcumin has been used in a wide range of clinical studies as a drug or adjuvant in the treatment of diseases, including those characterized by defective autophagy
[11]. Curcumin shows both activating
[13] and inhibitory
[4] actions on autophagy mechanisms. In particular, it regulates AMPK and can inhibit mTOR level/activity
[14]. The exact molecular mechanism by which curcumin exerts its role as an autophagy modulator remains to be elucidated; however, its activity as a potential therapeutic agent appears to be essential in different human disorders.
2. Autophagy: Mechanisms and Regulation
Autophagy maintains the cellular proteostasis by degrading misfolded and long-lived proteins. It also removes damaged organelles
[15]. It is divided into three subtypes according to the mechanism by which the intracellular material is destined to the lysosomes to be degraded: macroautophagy
[16], microautophagy
[17], and chaperone-mediated autophagy
[18] (
Figure 1).
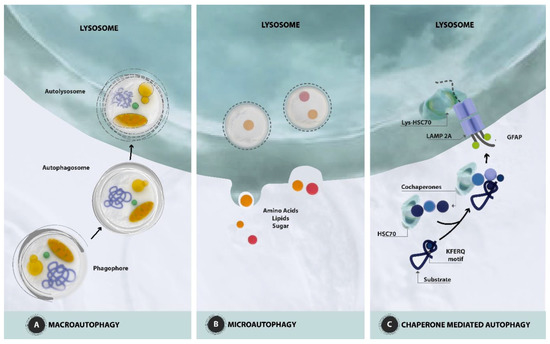
Figure 1. Schematic presentation of autophagic pathways. (A) Macroautophagy. The key event in macroautophagy is the de novo formation of a new organelle called the autophagosome, which surrounds and sequesters either random portions of the cytoplasm or selectively targets individual cytosolic components. Initially, double-membraned cup-shaped structures, called phagophores or membrane isolation, engulf the cytosolic cargo. The expansion of the double membrane ends with the completion of the autophagosome that is trafficked by microtubules. The fusion of the autophagosome with the lysosome constitutes an autolysosome where the trapped cargo can be degraded. (B) Microautophagy. The cytosolic cargo (amino acids, lipids, sugar) is translocated into the lysosomes for degradation via direct invagination, protrusion, or septation of the lysosomal limiting membrane. (C) Chaperone mediated autophagy. It selects a defined pool of proteins that contains the KFERQ motif and delivers them to lysosomes via the chaperone HSC70 and cochaperones. The complex formed by HSC70 and the KFERQ-containing protein interacts with the lysosome-associated surface membrane protein type 2A (LAMP2A). Then, the KFERQ-containing protein unfolds and is transported across the lysosome membrane by a lysosome form of HSC70 (lys-HSC70), which resides inside the lysosome. GFAP, glial fibrillary acidic protein.
These three subtypes show different mechanisms of cargo recognition and molecular support. However, all these subtypes have lysosomes as the final target for cargo digestion and product recycling. Microautophagy occurs when cytoplasmic material is introduced into lysosomes through direct invagination of the lysosomal membrane. Chaperone-mediated autophagy (CMA) is characterized by direct translocation into lysosomes for the degradation of proteins containing a defined pentapeptide pattern (KFERQ). Macroautophagy is characterized by the formation of double-membrane subcellular structures, called autophagosomes, which transport the degradable content from the cytoplasm and route them into lysosomes for degradation. Then, the cells reuse these products of degradation.
Macroautophagy initiates with the formation of the double membrane (phagophore), which is defined as nucleation. The phagophore derives from the plasma membrane, Golgi, endoplasmic reticulum or mitochondria
[19], and envelops misfolded proteins or damaged organelles. The expansion of the phagophore ends with the completion of the autophagosome. The fusion of the autophagosome with lysosome constitutes an autolysosome within which the enclosed material, known as autophagic cargo, is degraded
[20].
At the molecular level, macroautophagy is initiated by two major complexes: (a) the UN51-like Ser/Thr kinases (ULK) complex and (b) the class III phosphatidylinositol-3-kinase (PI3K) that are recruited to the phagophore assembly site (PAS)
[21]. The ULK complex includes the ULK1/2 family, the FAK family kinase interacting protein of 200 kDa (FIP200), and ATG13
[22]. The PI3K complex, also defined as the Beclin1 complex, contains vacuolar protein sorting 34 (Vps34), p15 (VPS15), Beclin1 (ATG6), and Barkor (ATG14)
[23]. Beclin1 localizes on the ER membrane and is regulated by the anti-apoptotic dimer BCL-2 and BCL-XL. When autophagy is activated, Beclin1 dissociates from the BCL-2 complex and coordinates with Vps34
[24]. Then, the bulk phosphatidylinositol 3-phosphate [PtdIns(3)P] is recruited on the surface of the phagophore
[5]. Two ubiquitin-like complexes are responsible for the extension and closure of the autophagosome. The first complex is initiated by the interaction of Atg7 with Atg5, which, in turn, binds covalently to Atg12
[25]. This complex interacts with Atg16 to form the Atg5-Atg12-Atg16 complex, which elongates the phagophore. The interaction between Atg9 with Atg2 and Atg18 is responsible for the trafficking between the Trans-Golgi Network, endosomes, and newly formed autophagosomes. Atg4B cleaves to the microtubule-associated protein 1 light chain 3 (LC3) in another ubiquitin-like complex, leading to the formation of LC3-I
[26]. Upon an autophagic signal, LC3-I is conjugated by Atg7, Atg3, and Atg12-Atg5-Atg16L multimers to a phosphatidylethanolamine (PE) moiety for the generation of the LC3-II form, which is considered a marker of autophagosome
[27]. Next, dynein and other motor proteins are involved in the transport of autophagosomes along the microtubules
[28]. Finally, the SNARE proteins (Soluble NSF Attachment Protein Receptors) are recruited on the lysosomes that fuse with the autophagosomes, leading to the degradation of the cargoes
[29].
Autophagy is induced by both physiological conditions and stress, such as hypoxia and food deprivation
[30]. Thus, different pathways regulate the autophagy machinery.
The IGF-1/Insulin pathways sense the nutrient variations and regulate growth, morphogenesis, and survival. In
C.elegans, a link has been shown between the IGF-1/Insulin pathway and autophagy. Mutants in certain autophagy genes affect IGF-1/Insulin-induced cell growth
[31]. In mammals, insulin withdrawal induces autophagic cell death in hippocampal neural stem cells
[32]. IGF-1/insulin, along with the mammalian target of rapamycin (mTOR), regulates autophagy at several levels. mTOR is a negative regulator of autophagy, which is modulated by proteins acting upstream to mTOR signaling
[33]. PTEN and TSC1/2 induce autophagy, whereas Akt inhibits it
[33]. Elongation factor-2, a downstream effector of mTOR, also modulates autophagy
[34]. The inhibitor of the mTOR complex 1 (MTORC1) induces the nuclear translocation of the transcription factor EB (TFEB) by phosphorylating key serine residues of TFEB, which in turn activates the expression of genes involved in autophagy and lysosomal biogenesis
[35][36]. The thioredoxin interacting protein (TXNIP), which modulates the cell and body metabolism and is down-regulated by insulin
[37][38][39], modulates autophagy through the mTOR pathway
[40]. In addition, TXNIP suppresses the activity of Atg4B, leading to activation of the autophagic flux
[41].
The tumor suppressor p53 modulates autophagy by regulating the expression of autophagy-related genes
[34]. P53 promotes autophagy by inducing the expression of the damage-regulated autophagy modulator (DRAM), which is an integral lysosomal membrane protein and assists in the accumulation of autophagosomes
[42]. P53 regulates autophagy by modulating the expression of chromatin-remodeling factors, such as e2f1
[43], which binds the gene regulatory regions of the autophagy proteins Atg1, Atg8, and DRAM
[44].
The transcription factors FOXO directly regulate the expression of Atg8 and Atg12
[45]. In both normal growth and starvation conditions, Sirt1 promotes autophagy by acetylating Atg5, Atg7, and Atg8
[46]. Reactive Oxygen Species (ROS) are metabolites produced by several cellular activities, mostly by respiration, and induce oxidative stress. The majority of ROS production occurs in mitochondria. ROS are very reactive species, and their deleterious effects are counteracted by autophagic degradation of mitochondria (mitophagy) and anti-oxidant defense
[47]. Indeed, ROS are necessary for the induction of autophagy and activate Atg4
[48]. Moreover, several stresses, such as hypoxia and exercise, induce ROS-dependent autophagy
[49].
CMA differs from the other two types of autophagy because it selects a defined pool of proteins that contains the KFERQ motif and delivers them to lysosomes. This peptide sequence is recognized by the heat shock protein of 70 kDa (hsc70), which is the chaperone targeting these proteins to lysosomes
[50]. The complex formed by hsc70 and the KFERQ-containing protein interacts with the lysosome-associated surface membrane protein type 2A (LAMP 2A). Then, the KFERQ-containing protein unfolds and is transported across the lysosome membrane by a lysosome form of hsc70 (lys-hsc70)
[50].
Autophagy typically enhances cell viability. However, alterations in its regulation are implicated in the pathogenesis of neurological and neuromuscular diseases. Indeed, it seems that induction of autophagy at early stages has a protective function against neurotoxicity
[51][52]. Because of its relevance in the pathogenesis of these diseases, it is the target of several pharmaceutical compounds. The characterization of natural compounds that target autophagy is an integral part of ongoing research aimed at finding therapeutic strategies for such diseases. In particular, recent studies emphasize the therapeutic challenges of curcumin in autophagy molecular mechanisms by highlighting curcumin as a potential therapeutic compound in neurological and neuromuscular diseases.
3. Curcumin Structure and Activity
Curcumin is a polyphenol derived from the turmeric root of Curcuma longa
[1]. Curcumin possesses two similar aromatic rings in which the o-methoxy phenolic groups are linked to an α,β-unsaturated β-diketone moiety
[53] (
Figure 2).
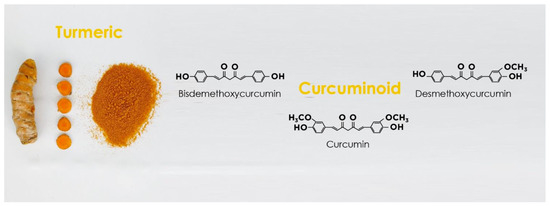
Figure 2. Curcuminoids in turmeric and their chemical structures.
Curcumin can also act as an electron donor in many redox reactions because it contains conjugated double bonds in its chemical structure, thereby stabilizing the structure
[53]. Notably, curcumin at a low concentration can act as an antioxidant agent and counteract the formation of oxidative stress because it functions as a scavenger of ROS
[54]. Oxidative stress is the result of an imbalance between the formation of ROS and the cellular antioxidant mechanisms, leading to peroxidation of membrane lipids and oxidative damage of proteins and DNA. Because of its structure, curcumin accumulates in hydrophobic regions, such as the plasma membrane, and decreases lipid peroxidation. In addition, curcumin stimulates antioxidant enzymes, such as catalase, superoxide dismutase (SOD), glutathione peroxidase (GPx), and heme oxygenase 1 (OH1), counteracting the oxidative damage
[55]. On the contrary, at higher concentrations, curcumin shows a pro-oxidant activity that promotes cancer cell apoptosis, playing an important function in cell death during the neoplastic process
[56]. Indeed, curcumin has a beneficial effect against cancer
[57][58][59].
Concerning the effect of oxidative stress on inflammation, several studies have investigated the effect of curcumin as an anti-inflammatory agent. These studies confirmed that curcumin inhibits inflammation by blocking several pro-inflammatory molecule (such as cyclooxygenase 2 (COX2) and lipoxygenase 5 (LOX5)), inducible nitric oxide synthase (iNOS), inflammatory cytokines (such as tumor necrosis factor α (TNFα)), interleukin-(IL-) 1, 2, 6, 8, and 12, monocyte chemoattractant protein 1 (MCP1), and transcription factors (such as activating protein 1 (AP1), and nuclear factor κB (NF-κB)
[59]. Thus, curcumin is believed to be beneficial against several diseases, including cancer, diabetes, and cardiovascular diseases
[55][59] as well as neurodegenerative and neuromuscular diseases, which we describe later in this review.
Curcumin is considered a beneficial treatment for several diseases because it confers health benefits and is well tolerated without any toxicity at high oral doses. It has been shown that patients tolerate up to 2.2 g of Curcuma extract containing 180 mg of curcumin/day for four months
[60]. Recently, we demonstrated that 1200 mg/day of curcumin for six months was well tolerated and reduced the size of tumors in neurofibromatosis type 1 patients
[57].
Also, curcumin is proposed as a therapeutic agent for brain tumors and neurodegenerative diseases, since it can cross the blood-brain barrier (BBB) after oral administration when it is in its native form (unglucuronidated and unsulfated)
[61].
Interestingly, curcumin is naturally fluorescent and binds misfolded proteins that are a pathogenic characteristic of several neurodegenerative diseases, such as the amyloid-beta aggregates in Alzheimer’s disease (AD)
[62]. For this reason, curcumin has been used to label the amyloid plaques
[62]. Moreover, curcumin can inhibit the aggregation of misfolded proteins and enhance their clearance
[61]. This function will be discussed in more detail below in this review.
Despite the beneficial effect of curcumin for health and the cure and prevention of several diseases, its clinical application shows limitations due to curcumin’s poor bioavailability. Indeed, oral treatment is affected by curcumin’s slow water solubility, poor absorption, rapid metabolism, and systemic elimination. For this reason, there are several studies aimed at improving the bioavailability of curcumin using different approaches: the use of adjuvants, such as piperine, quercetin, and resveratrol
[63]; nanoparticle-based delivery of curcumin using liposomes, solid lipid nanoparticles, niosomes, polymeric nanoparticles, polymeric micelles, cyclodextrins, dendrimers, and silver and gold nanoparticles
[64][65]. In addition, synthetic structural analogs of curcumin have been developed in order to improve its bioavailability
[66][67]. Interestingly, a recent study by our research group indicated that the bioavailability of curcumin significantly increases when its oral administration is associated with the Mediterranean Diet (MeDi)
[57][68]. These data suggest that the high concentration of extra virgin olive oil polyphenols and/or fatty acids present in the MeDi contributes to increasing the bioavailability of curcumin and enhancing its effects
[57].
 +1 point
+1 point
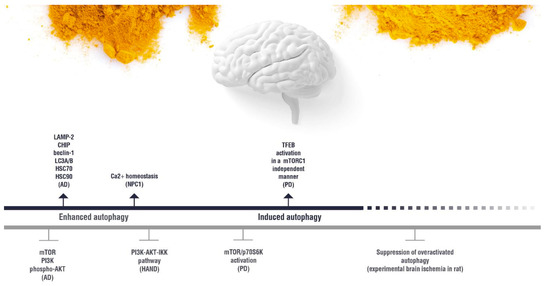
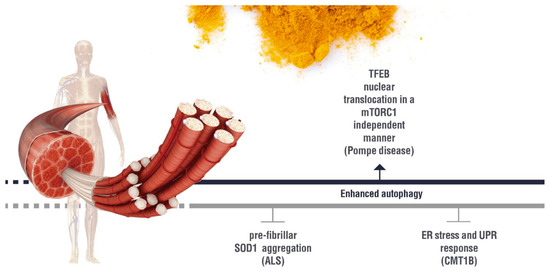
 (induction);
(induction);  (inhibition). AD: Alzheimer’s disease; HAND: HIV-induced neurocognitive disorder; PD: Parkinson’s disease; NPC1: Niemann Pick C1.
(inhibition). AD: Alzheimer’s disease; HAND: HIV-induced neurocognitive disorder; PD: Parkinson’s disease; NPC1: Niemann Pick C1.