 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christos Kyriakopoulos | + 6216 word(s) | 6216 | 2021-08-17 03:44:02 | | | |
| 2 | Christos Kyriakopoulos | Meta information modification | 6216 | 2021-08-17 18:07:08 | | | | |
| 3 | Rita Xu | -2440 word(s) | 3776 | 2021-08-18 03:54:22 | | |
Video Upload Options
Chronic Obstructive Pulmonary Disease (COPD) is a chronic inflammatory disease with multisystemic manifestations. Studies either held on stable disease patients or during exacerbations, have demonstrated that COPD is strongly related to venous thromboembolism and cardiovascular events.
1. Introduction
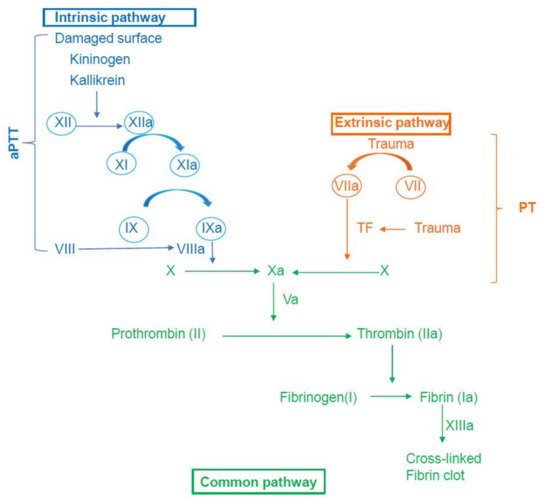
2. An Overview of the Coagulation Mechanism
3. Hypercoagulability in Stable COPD
4. Hypercoagulability during COPD exacerbations
| Author, Year | Participants | Disease Status | Results |
|---|---|---|---|
| Agale, 2018 [56] | 50 patients with stable COPD vs. 50 healthy controls | Stable COPD |
|
| Arregui, 2010 [57] | 51 patients with stable COPD vs. 30 healthy controls | Stable COPD |
|
| Ashitani, 2002 [8] | 40 patients with stable COPD vs. 20 healthy controls | Stable COPD |
|
| Cella, 2001 [62] | 14 patients with stable COPD vs. 20 healthy controls | Stable COPD |
|
| Daga, 2020 [75] | 30 patients with AECOPD vs. 30 stable COPD patients | AECOPD |
|
| Eickhoff, 2008 [54] | 60 patients with stable COPD vs. 20 healthy controls | Stable COPD |
|
| Elsalam, 2013 [74] | 38 patients with AECOPD vs. 25 healthy controls | AECOPD |
|
| 15 patients with severe COPD vs. 10 patients with moderate COPD | AECOPD |
|
|
| Garcia-Rio, 2010 [53] | 324 patients with stable COPD vs. 110 healthy controls | Stable COPD |
|
| Husebø, 2021 [76] | 413 patients with stable COPD vs. 49 healthy controls | Stable COPD |
|
| 148 COPD patients during AECOPD and compared to baseline values | AECOPD |
|
|
| Jankowski, 2011 [11] | 60 stable COPD patients | Stable COPD |
|
| Koutsokera, 2009 [68] | 30 COPD patients during AECOPD and compared to convalescence values (40 days after AECOPD) | AECOPD |
|
| Kyriakopoulos, 2020 [59] | 103 patients with stable COPD vs. 42 COPD-free smokers | Stable COPD |
|
| Maclay, 2011 [71] | 18 patients with stable COPD vs. 16 healthy controls | Stable COPD |
|
| 12 COPD patients during AECOPD and compared to convalescence values (2 weeks after AECOPD) | AECOPD |
|
|
| Polatli, 2008 [58] | 33 patients with stable COPD vs. 16 healthy controls | Stable COPD |
|
| 26 patients during AECOPD vs. 33 stable COPD patients | AECOPD |
|
|
| Roland, 1999 [77] | 30 COPD patients during AECOPD and compared to baseline values (4–6 weeks later) | AECOPD |
|
| Saldias, 2012 [67] | 85 COPD patients during AECOPD and compared to baseline values | AECOPD |
|
| Samareh, 2000 [55] | 31 patients with stable COPD vs. 29 healthy controls | Stable COPD |
|
| Silva, 2012 [78] | 58 patients with stable COPD vs. 30 healthy controls | Stable COPD |
|
| Song, 2013 [72] | 30 AECOPD patients with ARF II vs. 30 AECOPD patients without ARF II | AECOPD |
|
| Szczypiorska, 2015 [61] | 66 patients with stable COPD vs. 25 healthy controls | Stable COPD |
|
| Thomas, 2016 [70] | 20 patients with stable COPD vs. 20 healthy controls | Stable COPD |
|
| 20 patients during AECOPD vs. 20 stable COPD patients | AECOPD |
|
|
| Undas, 2011 [14] | 60 patients with stable COPD vs. 43 non-COPD smokers | Stable COPD |
|
| Vaidyula, 2009 [60] | 11 patients with stable COPD vs. 10 healthy controls | Stable COPD |
|
| Valipour, 2008 [69] | 30 patients with stable COPD vs. 30 healthy controls | Stable COPD |
|
| 30 COPD patients during AECOPD and compared to convalescence values (6 weeks after AECOPD) | AECOPD |
|
|
| 30 patients during AECOPD vs. 30 healthy controls | AECOPD |
|
|
| Van der Vorm, 2020 [73] | 52 COPD patients during AECOPD and compared to convalescence values * (8 weeks after AECOPD) | AECOPD |
|
| Waschki, 2017 [63] | 74 patients with stable COPD vs. 18 healthy controls | Stable COPD |
|
| Wedzicha, 2000 [34] | 67 COPD patients during AECOPD and compared to baseline values | AECOPD |
|
| Zhang, 2016 [12] | 43 patients with stable COPD vs. 43 healthy controls | Stable COPD |
|
| 43 COPD patients during AECOPD and compared to baseline values | AECOPD |
|
Values are presented as mean ± SD or median (IQR). § Without statistical significance. * Convalescence values available for 32 patients.
5. Conclusions
To the best of our knowledge, this is the first comprehensive review of studies on hypercoagulability either in stable COPD or during COPD exacerbations. A large body of epidemiological data supports the hypothesis that COPD is closely linked to a hypercoagulable state. Overall, patients with stable COPD exhibit major alterations of fibrinogen, FII, FV, FVII, FVIII, FIX, D-dimers, von Willebrand factor Ag, von Willebrand factor Ac, TF, TAT, FPA, β-thromboglobulin, tPA-PAI, prothrombin fragment 1 and 2, and maximum thrombin levels, pointing to hypercoagulability. Prothrombotic state is further enhanced during exacerbations, reflected by a significant increase of TF, TAT, soluble fibrin complex, fibrinogen, D-dimers, and APC-PCI. This summary provides an in‐depth overview on the alterations of coagulation factors and prothrombotic changes in COPD patients that enhance our understanding of the coexistence of cardiovascular comorbidities in these patients.
References
- Adeloye, D.; Chua, S.; Lee, C.; Basquill, C.; Papana, A.; Theodoratou, E.; Nair, H.; Gasevic, D.; Sridhar, D.; Campbell, H.; et al. Global and regional estimates of COPD prevalence: Systematic review and meta–analysis. J. Glob. Health 2015, 5, 020415.
- The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 28 May 2021).
- Schneider, C.; Bothner, U.; Jick, S.S.; Meier, C.R. Chronic obstructive pulmonary disease and the risk of cardiovascular diseases. Eur. J. Epidemiol. 2010, 25, 253–260.
- Chen, W.; Thomas, J.; Sadatsafavi, M.; FitzGerald, J.M. Risk of cardiovascular comorbidity in patients with chronic obstructive pulmonary disease: A systematic review and meta-analysis. Lancet Respir. Med. 2015, 3, 631–639.
- Lankeit, M.; Held, M. Incidence of venous thromboembolism in COPD: Linking inflammation and thrombosis? Eur. Respir. J. 2016, 47, 369–373.
- Sin, D.D.; Wu, L.; Paul Man, S.F. The relationship between reduced lung function and cardiovascular mortality. Chest 2005, 127, 1952–1959.
- Abduganieva, E.; Artikov, D.; Liverko, I. Hypercoagulation as the main cause of death in COPD. In Proceedings of the ERS International Congress 2019 Abstracts, Madrid, Spain, 28 September–2 October 2019.
- Ashitani, J.-I.; Mukae, H.; Arimura, Y.; Matsukura, S. Elevated plasma procoagulant and fibrinolytic markers in patients with chronic obstructive pulmonary disease. Intern. Med. 2002, 41, 181–185.
- Wanner, A.; Schmid, A. Faculty opinions recommendation of fibrin clot properties are altered in patients with chronic obstructive pulmonary disease. Beneficial effects of simvastatin treatment. Natl. Libr. Med. 2010, 102, 1176–1182.
- Sambola, A.; Osende, J.; Hathcock, J.; Degen, M.; Nemerson, Y.; Fuster, V.; Crandall, J.; Badimon, J.J. Role of risk factors in the modulation of tissue factor activity and blood thrombogenicity. Circulation 2003, 107, 973–977.
- Jankowski, M.; Undas, A.; Kaczmarek, P.; Butenas, S. Activated factor XI and tissue factor in chronic obstructive pulmonary disease: Links with inflammation and thrombin generation. Thromb. Res. 2011, 127, 242–246.
- Zhang, M.; Zhang, J.; Zhang, Q.; Yang, X.; Shan, H.; Ming, Z.; Chen, H.; Liu, Y.; Yin, J.; Li, Y. D-Dimer as a potential biomarker for the progression of COPD. Clin. Chim. Acta 2016, 455, 55–59.
- Zuur-Telgen, M.; van der Valk, P.; Zuur, B.; van der Palen, J.; Kerstjens, H.; Brusse-Keizer, M. Stable state fibrinogen is additive to Mr-proADM as a predictor of mortality in COPD patients. Eur. Res. J. 2015, 46, 3362.
- Undas, A.; Jankowski, M.; Kaczmarek, P.; Sladek, K.; Brummel-Ziedins, K. Thrombin generation in chronic obstructive pulmonary disease: Dependence on plasma factor composition. Thromb. Res. 2011, 128, e24–e28.
- Mispelaere, D.; Glerant, J.C.; Audebert, M.; Remond, A.; Sevestre-Pietri, M.A.; Jounieaux, V. Pulmonary embolism and sibilant types of chronic obstructive pulmonary disease decompensations. Rev. Mal. Respir. 2002, 19, 415–423.
- Kim, V.; Goel, N.; Gangar, J.; Zhao, H.; Ciccolella, D.; Silverman, E.; Crapo, J.; Criner, G. Risk Factors for venous thromboembolism in chronic obstructive pulmonary disease. Chronic Obstr. Pulm. Dis. 2014, 1, 239–249.
- Gan, W.Q. Association between chronic obstructive pulmonary disease and systemic inflammation: A systematic review and a meta-analysis. Thorax 2004, 59, 574–580.
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2017, 38, 785–791.
- Wang, Q.; Zennadi, R. Oxidative stress and thrombosis during aging: The roles of oxidative stress in RBCs in venous thrombosis. Int. J. Mol. Sci. 2020, 21, 4259.
- Bärtsch, P. How thrombogenic is hypoxia? JAMA 2006, 295, 2297–2299.
- Sabit, R.; Thomas, P.; Shale, D.J.; Collins, P.; Linnane, S.J. The effects of hypoxia on markers of coagulation and systemic inflammation in patients with COPD. Chest 2010, 138, 47–51.
- Lonergan, M.; Dicker, A.J.; Crichton, M.L.; Keir, H.R.; van Dyke, M.K.; Mullerova, H.; Miller, B.E.; Tal-Singer, R.; Chalmers, J.D. Blood neutrophil counts are associated with exacerbation frequency and mortality in COPD. Respir. Res. 2020, 21, 166.
- Iba, T.; Levy, J.H. Inflammation and thrombosis: Roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost. 2018, 16, 13911.
- Sin, D.D.; Man, S.F. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? the potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation 2003, 107, 1514–1519.
- Van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179.
- Gutmann, C.; Siow, R.; Gwozdz, A.M.; Saha, P.; Smith, A. Reactive oxygen species in venous thrombosis. Int. J. Mol. Sci. 2020, 21, 1918.
- Darbousset, R.; Thomas, G.M.; Mezouar, S.; Frère, C.; Bonier, R.; Mackman, N.; Renné, T.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Tissue factor–positive neutrophils bind to injured endothelial wall and Initiate thrombus formation. Blood 2012, 120, 2133–2143.
- Gupta, N.; Zhao, Y.-Y.; Evans, C.E. The stimulation of thrombosis by hypoxia. Thromb. Res. 2019, 181, 77–83.
- Mackman, N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arter. Thromb. Vasc. Biol. 2004, 24, 1015–1022.
- Prchal, J.T. Hypoxia and thrombosis. Blood 2018, 132, 348–349.
- Couturaud, F.; Bertoletti, L.; Pastre, J.; Roy, P.-M.; Le Mao, R.; Gagnadoux, F.; Paleiron, N.; Schmidt, J.; Sanchez, O.; De Magalhaes, E.; et al. Prevalence of pulmonary embolism among patients with COPD hospitalized with acutely worsening respiratory symptoms. JAMA 2021, 325, 59–68.
- Aleva, F.E.; Voets, L.W.L.M.; Simons, S.O.; de Mast, Q.; van der Ven, A.J.A.M.; Heijdra, Y.F. Prevalence and localization of pulmonary embolism in unexplained acute exacerbations of COPD: A systematic review and meta-analysis. Chest 2017, 151, 544–554.
- Polosa, R.; Malerba, M.; Cacciola, R.R.; Morjaria, J.B.; Maugeri, C.; Prosperini, G.; Gullo, R.; Spicuzza, L.; Radaeli, A.; Di Maria, G.U. Effect of acute exacerbations on circulating endothelial, clotting and fibrinolytic markers in COPD patients. Intern. Emerg. Med. 2013, 8, 567–574.
- Wedzicha, J.A.; Seemungal, T.A.; MacCallum, P.K.; Paul, E.A.; Donaldson, G.C.; Bhowmik, A.; Jeffries, D.J.; Meade, T.W. Acute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levels. Thromb. Haemost. 2000, 84, 210–215.
- Smeeth, L.; Cook, C.; Thomas, S.; Hall, A.J.; Hubbard, R.; Vallance, P. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet 2006, 367, 1075–1079.
- Schmidt, M.; Horvath-Puho, E.; Thomsen, R.W.; Smeeth, L.; Sørensen, H.T. Acute infections and venous thromboembolism. J. Intern. Med. 2012, 271, 608–618.
- Pottier, P.; Hardouin, J.B.; Lejeune, S.; Jolliet, P.; Gillet, B.; Planchon, B. Immobilization and the risk of venous thromboembolism. A meta-analysis on epidemiological studies. Thromb. Res. 2009, 124, 468–476.
- Dentali, F.; Pomero, F.; Micco, P.D.; La Regina, M.; Landini, F.; Mumoli, N.; Pieralli, F.; Giorgi-Pierfranceschi, M.; Re, R.; Vitale, J.; et al. Prevalence and risk factors for pulmonary embolism in patients with suspected acute exacerbation of COPD: A multi-center study. Eur. J. Intern. Med. 2020, 80, 54–59.
- Weitzenblum, E.; Chaouat, A.; Kessler, R.; Canuet, M. Overlap syndrome: Obstructive sleep apnea in patients with chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2008, 5, 237–241.
- Colten, H.R.; Altevogt, B.M. Sleep Disorders and Sleep Deprivation: An Unmet Public Health Problem; National Academies Press: Washington, DC, USA, 2006.
- Fernández-Bello, I.; Monzón, M.E.; García Río, F.; Justo, S.R.; Cubillos-Zapata, C.; Casitas, R.; Sánchez, B.; Jaureguizar, A.; Acuña, P.; Alonso-Fernández, A.; et al. Procoagulant state of sleep apnea depends on systemic inflammation and endothelial damage. Arch. Bronconeumol. 2020, 20, 30546–30549.
- Xu, J.; Wang, X.; Meng, F.; Zhao, T.; Tang, T.; Wu, W.; Wang, W. The role of obstructive sleep apnea on the prognosis of pulmonary embolism: A systemic review and meta-analysis. Sleep Breath. 2020, 24, 1–8.
- Xie, J.; Li, F.; Wu, X.; Hou, W. Prevalence of pulmonary embolism in patients with obstructive sleep apnea and chronic obstructive pulmonary disease: The overlap syndrome. Heart Lung 2019, 48, 261–265.
- Loeffen, R.; Spronk, H.M.H.; Ten Cate, H. The impact of blood coagulability on atherosclerosis and cardiovascular disease. J. Thromb. Haemost. 2012, 10, 1207–1216.
- Borissoff, J.I.; Spronk, H.M.H.; ten Cate, H. The hemostatic system as a modulator of atherosclerosis. N. Engl. J. Med. 2011, 364, 1746–1760.
- Ye, Z.; Liu, E.H.C.; Higgins, J.P.T.; Keavney, B.D.; Lowe, G.D.O.; Collins, R.; Danesh, J. Seven haemostatic gene polymorphisms in coronary disease: Meta-Analysis of 66,155 CASES and 91,307 controls. Lancet 2006, 367, 651–658.
- Elsevier Guyton and Hall Textbook of Medical Physiology. Available online: https://www.elsevier.com/books/guyton-and-hall-textbook-of-medical-physiology/hall/978-1-4557-7005-2 (accessed on 11 May 2021).
- Marder, V.J.; Aird, W.C.; Bennett, J.S.; Schulman, S.; White, G.C., II. Hemostasis and Thrombosis: Basic Principles and Clinical Practice; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; ISBN 9781451177695.
- Tapson, V.F. The role of smoking in coagulation and thromboembolism in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 71–77.
- Goldhaber, S.Z.; Tapson, V.F. A prospective registry of 5,451 patients with ultrasound-confirmed deep vein thrombosis. Am. J. Cardiol. 2004, 93, 259–262.
- Bertoletti, L.; Quenet, S.; Laporte, S.; Sahuquillo, J.C.; Conget, F.; Pedrajas, J.M.; Martin, M.; Casado, I.; Riera-Mestre, A.; Monreal, M. Pulmonary embolism and 3-month outcomes in 4036 patients with venous thromboembolism and chronic obstructive pulmonary disease: Data from the RIETE registry. Respir. Res. 2013, 14, 75.
- Mannino, D.M.; Ford, E.S.; Redd, S.C. Obstructive and restrictive lung disease and markers of inflammation: Data from the third national health and nutrition examination. Am. J. Med. 2003, 114, 758–762.
- Garcia-Rio, F.; Miravitlles, M.; Soriano, J.B.; Muñoz, L.; Duran-Tauleria, E.; Sánchez, G.; Sobradillo, V.; Ancochea, J. EPI-SCAN steering committee systemic inflammation in chronic obstructive pulmonary disease: A population-based study. Respir. Res. 2010, 11, 63.
- Eickhoff, P.; Valipour, A.; Kiss, D.; Schreder, M.; Cekici, L.; Geyer, K.; Kohansal, R.; Burghuber, O.C. Determinants of systemic vascular function in patients with stable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 178, 777–780.
- Samareh, M.F.; Khorasani, S.B.R.; Shadkam, S. Correlation of CRP and serum fibrinogen levels with disease severity, clinical factors and pulmonary function tests in COPD patients. Tanaffos 2010, 9, 28–33.
- Agale, S.A. Serum fibrinogen level in COPD patients—A comparative study. J. Med. Sci. Clin. Res. 2018, 6, 701–703.
- Arregui, M.A.; Ezquerra, K.L.; López, F.C.; Lacasa, R.C. Hypercoagulability state and endotelial injury in stable chronic obstructive pulmonary disease patients. An. Sist. Sanit. Navar. 2010, 33, 43–50.
- Polatli, M.; Cakir, A.; Cildag, O.; Bolaman, A.Z.; Yenisey, C.; Yenicerioglu, Y. Microalbuminuria, von willebrand factor and fibrinogen levels as markers of the severity in COPD exacerbation. J. Thromb. Thrombolysis 2008, 26, 97–102.
- Kyriakopoulos, C.; Chronis, C.; Tatsioni, A.; Papapetrou, E.; Gogali, A.; Vaggeli, K.; Katsanos, C.; Tsaousi, C.; Tsalepi, C.; Charisis, A.; et al. Hypercoagulability in patients with stable COPD assessed by coagulation factor levels. Eur. Respir. J. 2020, 56 (Suppl. 64), 3578.
- Vaidyula, V.R.; Criner, G.J.; Grabianowski, C.; Rao, A.K. Circulating tissue factor procoagulant activity is elevated in stable moderate to severe chronic obstructive pulmonary disease. Thromb. Res. 2009, 124, 259–261.
- Szczypiorska, A.; Czajkowska-Malinowska, M.; Góralczyk, B.; Bielis, L.; Drela, E.; Góralczyk, K.; Ruszkowska-Ciastek, B.; Rość, D. Tissue factor and tissue factor pathway Inhibitor in chronic obstructive pulmonary disease. Med. Res. J. 2015, 3, 32–37.
- Cella, G.; Sbarai, A.; Mazzaro, G.; Vanzo, B.; Romano, S.; Hoppensteadt, T.; Fareed, J. Plasma markers of endothelial dysfunction in chronic obstructive pulmonary disease. Clin. Appl. Thromb. Hemost. 2001, 7, 205–208.
- Waschki, B.; Watz, H.; Holz, O.; Magnussen, H.; Olejnicka, B.; Welte, T.; Rabe, K.F.; Janciauskiene, S. Plasminogen activator inhibitor-1 is elevated in patients with COPD independent of metabolic and cardiovascular function. Int. J. Chron. Obstr. Pulm. Dis. 2017, 12, 981–987.
- Pang, H.; Wang, L.; Liu, J.; Wang, S.; Yang, Y.; Yang, T.; Wang, C. The prevalence and risk factors of venous thromboembolism in hospitalized patients with acute exacerbation of chronic obstructive pulmonary disease. Clin. Respir. J. 2018, 12, 2573–2580.
- Zielinski, J.; MacNee, W.; Wedzicha, J.; Ambrosino, N.; Braghiroli, A.; Dolensky, J.; Howard, P.; Gorzelak, K.; Lahdensuo, A.; Strom, K.; et al. Causes of death in patients with COPD and chronic respiratory failure. Monaldi Arch. Chest Dis. 1997, 52, 43–47.
- Kim, T.H.; Oh, D.K.; Oh, Y.-M.; Lee, S.W.; Do Lee, S.; Lee, J.S. Fibrinogen as a potential biomarker for clinical phenotype in patients with chronic obstructive pulmonary disease. J. Thorac. Dis. 2018, 10, 5260–5268.
- Saldías, P.F.; Díaz, P.O.; Dreyse, D.J.; Gaggero, B.A.; Sandoval, A.C.; Lisboa, B.C. Etiology and Biomarkers of Systemic Inflammation in Mild to Moderate COPD Exacerbations. Rev. Med. Chil. 2012, 140, 10.
- Koutsokera, A.; Kiropoulos, T.S.; Nikoulis, D.J.; Daniil, Z.D.; Tsolaki, V.; Tanou, K.; Papaioannou, A.I.; Germenis, A.; Gourgoulianis, K.I.; Kostikas, K. Clinical, functional and biochemical changes during recovery from COPD exacerbations. Respir. Med. 2009, 103, 919–926.
- Valipour, A.; Schreder, M.; Wolzt, M.; Saliba, S.; Kapiotis, S.; Eickhoff, P.; Burghuber, O.C. Circulating vascular endothelial growth factor and systemic inflammatory markers in patients with stable and exacerbated chronic obstructive pulmonary disease. Clin. Sci. 2008, 115, 225–232.
- Thomas, B.; Yuvarajan, S. Plasma fibrinogen in chronic obstructive pulmonary disease—A cross sectional study conducted in a tertiary care hospital in puducherry, India. Br. J. Med. Med. Res. 2016, 14, 1–8.
- Maclay, J.D.; McAllister, D.A.; Johnston, S.; Raftis, J.; McGuinnes, C.; Deans, A.; Newby, D.E.; Mills, N.L.; MacNee, W. Increased platelet activation in patients with stable and acute exacerbation of COPD. Thorax 2011, 66, 769–774.
- Song, Y.-J.; Zhou, Z.-H.; Liu, Y.-K.; Rao, S.-M.; Huang, Y.-J. Prothrombotic state in senile patients with acute exacerbations of chronic obstructive pulmonary disease combined with respiratory failure. Exp. Ther. Med. 2013, 5, 1184–1188.
- Van der Vorm, L.N.; Li, L.; Huskens, D.; Hulstein, J.J.J.; Roest, M.; de Groot, P.G.; Ten Cate, H.; de Laat, B.; Remijn, J.A.; Simons, S.O. Acute exacerbations of COPD are associated with a prothrombotic state through platelet-monocyte complexes, endothelial activation and increased thrombin generation. Respir. Med. 2020, 171, 106094.
- Elsalam, H.M.A.; Mohamed, M.A.; El Gammal, M.S.; El-Shabrawy, M. Hypercoagulability in different respiratory diseases. Egypt. J. Chest Dis. Tuberc. 2013, 62, 331–341.
- Daga, M.K.; Kumar, N.; Mawari, G.; Singh, S.; Mahapatra, S.J.; Hussain, M.; Jha, M.K. Study of prothrombotic markers in COPD. J. Adv. Res. Med. 2020, 7, 1–6.
- Husebø, G.R.; Gabazza, E.C.; D’Alessandro Gabazza, C.; Yasuma, T.; Toda, M.; Aanerud, M.; Nielsen, R.; Bakke, P.S.; Eagan, T.M.L. Coagulation markers as predictors for clinical events in COPD. Respirology 2021, 26, 342–351.
- Roland, M.A.; Seemungal, T.A.R.; Bhowmik, A.; Sapsford, R.J.; Wedzicha, J.A.; MacCallum, P.K. P17 Effect of exacerbation on plasma levels of tissue factor in patients with COPD. Blood Coagul. Fibrinolysis 1999, 10, 544.
- Silva, D.R.; Coelho, A.C.; Gazzana, M.B.; Menna Barreto, S.S.; Knorst, M.M. D-Dimer levels in stable COPD patients: A case-control study. Chronic Obstr. Pulm. Dis. 2012, 9, 426–431.


