1000/1000
Hot
Most Recent
 +1 point
+1 point
The immunobullous condition Pemphigus Vulgaris (PV) is caused by autoantibodies targeting the desmosomal adhesion proteins, leading to blistering in the skin and mucosal membrane. There is still no cure to the disease apart from the use of corticosteroids and immunosuppressive agents.
The autoimmune disorder, Pemphigus is a blistering disease that results in intraepithelial blisters affecting predominantly the mucosa and skin. It is mediated by circulating pathogenic autoantibodies that attack the keratinocyte cell surface proteins leading to acantholysis [1]. Acantholysis indicates the loss of intercellular connection between keratinocytes in the epidermal stratum spinosum that renders the intraepidermal clefts. Two common subtypes of pemphigus with distinct clinical and immunopathological features are characterized, i.e. Pemphigus Vulgaris (PV) and Pemphigus Foliaceus (PF) with PV comprises of approximately 70% of the pemphigus cases. Furthermore, there are two subsets of PV; the mucosal dominant form is caused by autoantibodies targeting desmoglein-3 (DSG3) only and the mucocutaneous form has the autoantibodies targeting both desmoglein-1 (DSG1) and DSG3.
Clinically, PV presents as multiple flaccid blisters limited to the suprabasal layer in the epidermis or mucosal membrane, whilst the keratinocytes in the top layers maintain their cell cohesion. Interestingly, the basal cells also maintain their adhesion to the basement membrane, thereby histologically appearing like a ’row of tombstone’ [2]. On the other hand, in PF, autoantibodies target DSG1 that leads to acantholysis in the granular layer of the epidermis, without affecting the oral mucosa, and clinically presents as superficial frail blisters [3][4]. If left untreated, PV is fatal due to the failure of the skin barrier that culminates in fluid loss, electrolyte disturbances and contracting the opportunistic infection. The most common treatment modality is the administration of oral steroids and immunosuppressive agents and so far, there is still a lack of specific treatment for these diseases.
The desmosomal cadherin DSG3 was first identified as an isoform of the desmoglein subfamily and the auto-antigen in PV [5]. Following this pioneer finding, the pathogenicity of anti-DSG3 autoantibodies in PV has been confirmed by ample in vivo and in vitro studies [6][7]. DSG3 also has been indicated as a key mediator involved in desmosome remodelling, epidermal proliferation and differentiation, cell migration and apoptosis, and thus validating that DSG3 acts as a signalling molecule that has a major impact on tissue integrity and homeostasis. The binding of PV IgG to DSG3 activates several signalling pathways, such as phospholipase C, p38 MAPK, Src and Rho A [8][9][10][11], leading to DSG3 depletion from the desmosome and cell surface. As a result, the loss of intercellular cohesion and the formation of blisters occur. The distribution of DSG3 is not only restricted to desmosomes but also is located at the plasma membrane. Therefore, it was thought that the non-junctional pool of DSG3 may be responsible for cell signalling and is the primary target of PV IgG [12]. In line with this notion, our previous study showed that non-junctional DSG3 regulates Src via interaction with classical E-cadherin and helps regulate the adherens junction formation [13][14], suggesting the DSG3’s broader role in epithelial cell biology. This entry summarises recent advances in our understanding of pemphigus pathology with a focus on new findings of DSG3’s signalling roles in the regulation of p53 and YAP that have important implications in pemphigus acantholysis [15][16].
Ample studies support the concept that PV is caused by the pathogenic IgG autoantibodies directed against DSG3 as well as DSG1 [5][17][18]. However, there is evidence also suggesting pemphigus blistering results from intracellular signalling mediated by non-DSG targeting autoantibodies [9][19][20][21][22]. Nevertheless, the pathological mechanisms of blister formation in PV remain not fully understood [20][23]. Our recent studies have identified two fundamental signalling pathways, i.e. p53 and YAP that are regulated by DSG3 and PV IgG targeting DSG3 causes dysregulation of both pathways, resulting in apoptosis and disruption of cell junctions.
Early studies have proposed that apoptosis may be responsible for pemphigus acantholysis [36,37]. A new terminology ‘apoptolysis’- basal cell shrinkage hypothesis was proposed by Grando et al. in 2009, to explain the pathogenesis of acantholysis. After the pathogenic PV autoantibodies bind to the keratinocyte receptor(s), a series of signal-transduction pathways trigger the rupture of the cytoskeleton, resulting in the collapse and shrinkage of the keratinocytes. This theory vividly links to cell apoptosis and basic pathological features, emphasizing apoptotic enzymes contribute to acantholysis development in terms of both molecular events and chronologic sequence [21].
Our recent study has identified DSG3 acting as an anti-stress protein by counterbalancing p53 in the maintenance of normal epithelial homeostasis and demonstrated disruption of this regulation in PV [15] (Figure 1). The study addresses the long-term controversy in the field of pemphigus research and fills the gap for the link between DSG3 and p53. It indicates that the loss/disruption of DSG3 alone is sufficient to induce aberrant p53 response and thus, the disruption of DSG3 mediated intercellular junctions does not result solely from steric hindrance but rather causes dysfunction of cellular anti-stress response. This finding uncovers a previously unidentified biological function of this desmosomal cadherin in keratinocytes and suggests that blisters are partly due to the altered stress response that acts in the orchestra with p53 resulting in the damage of normal tissue homeostasis. Hence, disruption of this pathway may play an important part in PV pathology.
Some signalling pathways that are activated in PV [9][24][25] are the known elements in the p53 signalling network such as p38 MAPK and c-Myc [26][27]. Thus, both p38 MAPK and c-Myc activation could participate in the DSG3/p53 pathway leading to apoptosis and blistering in PV. c-Myc is also known to regulate apoptosis via post-translational regulation of p53 stability [28]. Interestingly, Pusapati et al. demonstrated that the p53 induction in response to c-Myc overexpression requires ATM kinase, a major regulator of the cellular response to DNA double-strand breaks (DSBs) [29]. The study by our group showed evidence of DSBs and upregulation of ATM kinase in DSG3 depleted cells, along with upregulation of the p53 pathway [15].
Increased accumulation of reactive oxygen species (ROS) or free radicals can lead to oxidative stress. Although ROS is indispensable at moderate concentrations to defend from microorganisms [66], excess ROS can lead to opposite detrimental effects such as DNA damage, lipid peroxidation, dysregulated signal transduction, altered cellular function and even cell death [67,68]. DNA damages such as double-strand breaks (DSBs) are extremely toxic to cells and can cause diverse human diseases including cancer, due to p53 induction [29]. Antioxidants are the key components in this system and protect the cells against the toxic effects of excess free radicals. The imbalance of oxidant/antioxidant is identified in various autoimmune diseases including rheumatoid arthritis and lupus erythematosus [69–74]. However, there remain limited studies regarding oxidative stress in PV and in keratinocytes treated with PV IgG though the evidence obtained suggests this is the case [30][31][32][33][34][35]. Reduction of antioxidants and increased oxidative stress is found to be correlated to clinical disease activity [30][34]. However, it remains not completely clear whether the oxidant/antioxidant disequilibrium is the cause or effect of PV IgG-mediated disruption of cell-cell adhesion.
A recent study by our group has demonstrated that elevated ROS production can be evoked by treating keratinocytes with PV patients’ sera as well as a pathogenic mouse monoclonal antibody AK23, with concomitant accumulation of Yes-associated protein (YAP) especially in the cytoplasm of antibody-treated cells [16]. Importantly, a marked increase of YAP was also detected in the oral mucous membrane of PV patients with elevated YAP in cells not only surrounding blisters but in the perilesional regions. YAP is a crucial downstream co-transcription factor in the Hippo pathway and also is involved in the cellular anti-stress response in Hippo-dependent and –independent manners. Elevated YAP was demonstrated in cells exposed to hydrogen peroxide (H2O2) which mimics cellular oxidative stress. Enforced expression of exogenous YAP led to disruption of cell junctions (such as α-catenin), whereas an adverse effect was observed in cells with YAP silencing with concomitantly enhanced DSG3 expression that was capable of abrogating AK23-mediated pathogenicity to DSG3 [16]. Significantly, our study showed that treating cells with antioxidants, such as glutathione (GSH) and N-acetyl cysteine (NAC), as well as the inhibitors for various MAPK pathways including p38 MAPK, was able to suppress H2O2-induced YAP coupled with enhanced cell-cell adhesion. These findings collectively suggest that antibody-induced oxidative stress and cytoplasmic YAP accumulation may attribute to the disruption of cell junctions and pemphigus acantholysis.
Overall, these findings indicate that PV autoantibodies targeting DSG3 affect its function in the anti-stress network that leads to oxidative/antioxidative imbalance in cells. As a consequence, oxidative stress can occur in PV cells resulting in DSBs and activation of a chain of reactions including ATM, c-Myc and p38 MAPK, and subsequently, the p53 pathway leading to deregulation of the apoptotic machinery and PV blistering (Figure 1).
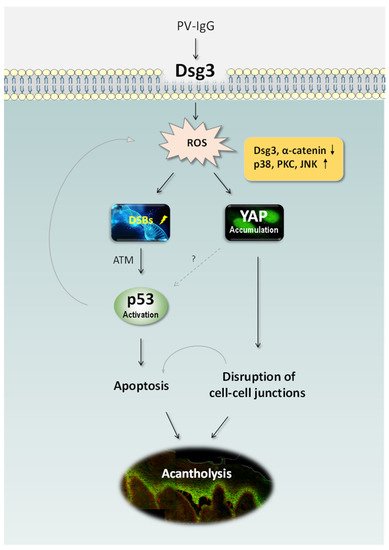
The pathological mechanisms underlying the pemphigus acantholysis are complex and remain not completely clear. Over the past three decades, significant advances have been made in our understanding of PV pathophysiology. Autoantibodies targeting the N-terminus of DSG3 are likely the initial and critical step that has an impact on its action in anti-stress response and hence triggers various intracellular events leading to blistering in DSG3 baring tissues such as the skin and oral mucosa. The direct inhibition of DSG3′s trans-interactions is not sufficient to induce the complete loss of cell cohesion and the synergistic action of orchestra signalling networks are considered to be vital in the pathophysiology of PV. Likely, the non-DSG antibodies also play a role [36][37]. Additionally, genetic predisposition to PV is well-reported and T cells are demonstrated as a critical player in autoimmunity. Furthermore, an increased incidence of apoptotic occurrence has been implicated in PV acantholysis lesions as elevated apoptotic factors such as Bax, FasL, and caspases as well as downregulation of the apoptotic inhibitory protein Bcl-2 in PV have been reported. Our recent studies shed new light on the unprecedented roles of DSG3 and suggest its mediated signalling towards p53, YAP and ROS in cellular stress response with the evidence of dysregulation of these axes being demonstrated in PV [15][16][38][39][40]. Therefore, elucidation of the underlying mechanisms could be a fertile area for future research. Moreover, the finding of oxidative stress-mediated YAP dysregulation in PV [16] opens a new avenue to explore a novel therapeutic approach with antioxidants.





