1000/1000
Hot
Most Recent
 +1 point
+1 point
The coronavirus disease 2019 (COVID-19) has spread over the world for more than one year. COVID-19 often develops life-threatening hypoxemia. Endothelial injury caused by the viral infection leads to intravascular coagulation and ventilation-perfusion mismatch, alveolar edema also plays a key role in the disease progression. Diffuse alveolar damage (DAD) is the histopathological pattern commonly described in COVID-19. Endothelial barrier disruption induces interstitial flooding via activation of the actin-myosin contractile apparatus. Then alveolar edema leads to hypoxia at the injured alveolar units. Hypoxia in turn inhibits edema fluid clearance, due in part to the disassembly of the keratin intermediate filament network, a fundamental element of the cellular cytoskeleton, therefore destructing the epithelial barrier. Therefore, a long-term hypoxia aggravates the disease by inducing more alveolar edema, which forms a vicious circle.
The lung’s initial response to acute viral infections has been characterized by innate immunity-mediated damages of the alveolar endothelial and epithelial barriers and accumulation of protein-rich edema fluid within the interstitium and alveolus, and then a great decline in oxygen diffusion over the blood-air barrier [1]. Blood flow through severely-damaged units, hereby, constitutes an intrapulmonary shunt and the hypoxia [1]. Acute hypoxia inhibits Na/K-ATPase function by activating its endocytosis from the plasma membrane to intracellular compartments. The endocytosis process is suggested to be mediated by the accumulation of reactive oxygen species (ROS) in mitochondria. Then ROS promotes the protein kinase C (PKC)-zeta dependent phosphorylation of the Na/K-ATPase α subunit triggering its endocytosis in a clathrin-AP2 dependent pathway [2]. More prolonged hypoxia may cause the ubiquitination and degradation of Na/K-ATPase subsequently. As a result, hypoxia inhibits K+ channels but activates voltage gated Ca2+ channels, which raises cytosolic Ca2+ levels in pulmonary artery smooth muscle cells (PASMC) and causes vasoconstriction [1][3]. Thus at the tissue level, patients of acute lung injury often develop the hypoxic pulmonary vasoconstriction (HPV), which is an essential protection mechanism of the lung that directs blood perfusion from badly-ventilated to well-ventilated alveoli to optimize gas exchange [4][5]. Endothelin-1 and thromboxane A2 may amplify, whereas prostacyclin and nitric oxide (NO) may moderate this process [5]. In addition, prostacyclin was suggested to be coupled mainly to cyclooxygenase-1 in acute hypoxia, but to cyclooxygenase-2 in chronic hypoxia [5].
At the subcellular level, mitochondria in PASMC have been considered to be oxygen sensors and initiate HPV [6][7][8][9][10]. Warburg metabolism (a phenomenon firstly found in tumor cells, which almost exclusively use glycolysis to generate energy, even under aerobic conditions) in PASMC mitochondria is initiated by the induction of a pseudo-hypoxic state, where DNA methyltransferase-mediated regulation in redox signaling results in normoxic activation of hypoxia inducible factor-1α (HIF-1α) and pyruvate dehydrogenase kinase (PDK) accumulation [6][7][8][9][10]. On the other hand, mitochondrial division has been also proved to be coordinated with nuclear division via the cellular process named mitotic fission. Increased mitotic fission in HPV, driven by enhanced fission but repressed fusion, accelerates the cell cycle and increases apoptosis resistance [6][7][8][9][10]. Thus, Warburg metabolisms sustain energy homeostasis through inhibiting oxidative metabolism, which may reduce mitochondrial apoptosis, and lead to uncontrolled cell division. Mitochondrial dynamic and metabolic disorders combine to show the hyper-proliferative and apoptosis-resistant phenotypes in pulmonary artery smooth muscle cells [6][7][8][9][10].
The renin-angiotensin-aldosterone system (RAAS) also plays a key role in endothelial injuries. RAAS is a key regulatory system of electrolyte homeostasis and pulmonary artery status and functions through angiotensin-converting enzyme (ACE)/angiotensin II (Ang II)/Ang II type 1 (AT1) receptor axis and angiotensin-converting enzyme 2 (ACE2)/angiotensin-1–7 Ang-(1–7)/MAS receptor axis. RAAS dysfunction has been proved to be related to the occurrence and development of acute pulmonary injuries and ARDS and may cause a serious prognosis and even death [11][12][13][14][15]. A large number of studies suggest that ACE2 works as the cellular receptor for SARS-CoV-2 entry [16][17][18]. Similar to other families of Coronaviridae viruses, the protein interactions between the virus and the membrane-bound ACE2 requires the cleavage of the spike glycoprotein S into two subunits. After the binding of the S1 receptor-binding domain (RBD) to ACE2, the trans-membrane protease/serine subfamily 2 (TMPRSS2) cleaves the S2 subunit and facilitates membrane fusion [16][17][18]. It has been confirmed that SARS-CoV-2 cell entry results in decline of the ACE2 protein, which converts Ang II into Ang-(1–7). Ang II induces pro-inflammatory cascades when binding to the AT1 receptor, while Ang-(1–7) generates anti-inflammatory effects through the interaction with the MAS receptor. Thus, down-regulation of ACE2 would result in pulmonary injury and vasoconstriction [11][12][13][14][15][19]. Besides these mechanisms, relative ACE2 deficiency may also lead to enhanced and protracted tissues, and vessel exposure to Ang II, which then enhances thrombosis, cell proliferation and recruitment, increases tissue permeability, cytokine production and results in inflammation (Table 1) [19][20].
Table 1. Pulmonary pathological changes in COVID-19.
| Injuries | Pathogenic Mechanisms | Refs. |
|---|---|---|
| Alveolar endothelial injury | Endothelial barrier disruption induces intrapulmonary shunt, hypoxia, intravascular coagulation and the release of pro-inflammatory factors. | [21][1][22] |
| ACE2-decline-induced pulmonary injury | ACE2 deficiency leads to enhanced and protracted tissues, and vessel exposure to Ang II, which then enhances thrombosis and cell proliferation, increases tissue permeability, cytokine production and inflammation. | [11][12][13][14][15][16][17][18][19][20] |
| Loss of hypoxic pulmonary vasoconstriction (HPV) | HPV directs blood perfusion from badly-ventilated to well-ventilated alveoli to optimize gas exchange. | [23][24][25] |
| General pulmonary vasoconstriction | Lead to pulmonary hypertension and a risk of right-heart failure subsequently. | [26][27][28] |
| Severe ventilation-perfusion mismatch | Induce hypoxemia in the non-injured fraction or/and cause hyper-perfusion of the small injured fraction. | [29][30] |
| Intravascular coagulation and microthrombi formation | Lead to increased wasted ventilation and less efficient carbon dioxide removal. | [31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49] |
| Diffuse alveolar damage (DAD) | Lead to hypoxia at the edematous alveoli. | [50][51][52] |
| Alveolar edema | Lead to great decline in oxygen diffusion over the blood-air barrier (hypoxia); Hypoxia in turn inhibits oedema fluid clearance. | [1][53] |
| Impaired alveolar surfactant production | Increase alveolar surface tension and hamper alveolar fluid resorption. | [54][55][56][57][58] |
HPV is thought to optimize gas exchange through shunting blood from poorly ventilated areas to those rich in oxygen. However, a general pulmonary vasoconstriction happens in both normoxic lung competencies and hypoxic lung competencies, which may lead to the pulmonary hypertension (PH) and a risk of right-heart failure subsequently, just as observed in patients with acute altitude sickness [5][22][23]. HPV has been observed in COVID-19 patients [24][25]. Moreover, the pulmonary hypertension and the subsequent right ventricular dysfunction also have been confirmed in severe COVID-19 patients (Table 1) [26][27]. Significant alternations in monocyte size, lymphocyte stiffness, neutrophil size and deformability, and heterogeneity of erythrocyte size and deformation in COVID-19 patients also indicated great changes in pulmonary blood stream dynamics [28].
Nevertheless, drugs to inhibit HPV, such as acetazolamide, calcium channel blockers and phosphodiesterase-5 inhibitors, should be avoided for COVID-19 patients, given that the consequences of COVID-19 may be exacerbated by loss of HPV, whether because of the destructive effects of the virus on mitochondria or the ability of endotoxin and inflammatory stimuli to eliminate HPV [25]. HPV attenuating drugs may exacerbate hypoxemia in COVID-19 pneumonia [25].
In some cases, dysregulated HPV may also cause mismatched blood flow and alveolar ventilation, which may restore the oxygen supply to the poorly-ventilated alveolar cells and result in life-threatening hypoxemia [22]. Recently, Herrmann et al. [29] modeled lung perfusion abnormalities and suggested that early COVID-19 hypoxemia may be mainly attributed to severe ventilation-perfusion mismatch. Their model predicted that calculated shunt fractions in excess of three times the injured fractions, which could be explained by (a) extensive perfusion defect, (b) perfusion defect combined with ventilation-perfusion mismatching in the non-injured fraction, or (c) hyper-perfusion of the small injured regions, with up to 3-fold increases in regional perfusion to the afflicted fraction [29]. Clinical features of COVID-19 pneumonia also confirmed the malfunction of oxygen-sensing responses including the ventilation-perfusion mismatch (Table 1) [30].
Pulmonary intravascular coagulation also plays an important role in the disease progression of COVID-19. After alveolar injuries, resident alveolar macrophages are activated, causing the release of potent proinflammatory mediators and chemokines that promote the accumulation of neutrophils and monocytes, such as vascular endothelial growth factor (VEGF), angiotensin II (Ang II), glycosaminoglycans (GAGs), von Willebrand factor (vWF) and soluble intercellular adhesion molecule (sICAM-1) [1]. Activated neutrophils further contribute to injuries through releasing toxic mediators. On the other hand, intravascular coagulation leads to platelet aggregation and micro-thrombi formation, which may aggravate the pulmonary injuries [1]. Intravascular coagulation primarily leads to increased dead space (increased wasted ventilation and less efficient carbon dioxide removal). Whereas inflammatory mediators from endothelial injury may worsen hypoxemia through exacerbate ventilation-perfusion mismatching (Table 1) [22][31]. Coagulation and thrombosis have been identified clinically as prominent symptoms of COVID-19 [32][33].
Nevertheless, neither intravascular coagulation nor ventilation-perfusion mismatch could be easily corrected. Oxygen supply is a common treatment to COVID-19 patients. Increasing inspired oxygen results in enhanced oxygenation but however does not improve the ratio of arterial oxygen tension to fractional inspired oxygen (PaO2:FiO2) [29]. Inhaled nitric oxide decreases total pulmonary vascular resistance [34][35] but however may increase blood flow to these low ventilation-perfusion regions, which causes further arterial desaturation (Table 2) [22].
Table 2. Putative drugs and treatments to SARS-CoV-2 pneumonia.
| Drugs or Treatments | Therapeutic Mechanisms | Therapeutic Effects | Refs. |
|---|---|---|---|
| Oxygen inhalation | Oxygenation enhancement | Alleviate hypoxia; however, high-flow oxygen lead to pulmonary toxic effects. | [22] |
| Mechanical ventilation | Oxygenation enhancement | Alleviate hypoxia; however, mechanical ventilation may cause mucus to be blown deep into the small airways, which then aggravates alveolar hypoxia. | [59] |
| ECMO | Oxygenation enhancement | Alleviate hypoxia; however, patients receiving ECMO still showed a high mortality rate. | [59] |
| Inhaled nitric oxide | Decrease pulmonary vascular resistance | Improve ventilation-perfusion ratio; however, may cause further arterial desaturation (hypoxia). | [22][34][35] |
| Aspirin | Anticoagulation | Reduce ICU cases, but show no apparent association with the fatality. | [36][37][38] |
| Heparin | Anticoagulation; block virus entry; increase antibody titres | Reduce the risk of in-hospital mortality and decrease the occurrence of severe cases; however, could not completely prevent occurrence of severe cases. | [39][40][41][42][43][44][45][46][47][48][49] |
| Exogenous pulmonary surfactant | Alleviate alveolar edema | Reduce the mortality of infants with neonatal RDS; however, clinical outcomes for COVID-19 patients need further investigation. | [54][55][56][57][58] |
| Ethanol–oxygen vapor therapy | Alleviate alveolar edema | May reduce occurrence of severe cases and the mortality rate (need clinical verification). | [60][61][62][63] |
| Conservative fluid management | Alleviate alveolar edema | May reduce occurrence of severe cases and the mortality rate (need clinical verification). | [64][65] |
Besides above pathogenic mechanisms, alveolar edema also plays a key role in the disease progression. Diffuse alveolar damage (DAD) is the histopathological pattern commonly described in COVID-19 (Table 1) [50][51][52]. Endothelial barrier disruption induces interstitial flooding via activation of the actin-myosin contractile apparatus [1]. Then alveolar edema leads to hypoxia at the injured alveolar units [1]. Hypoxia in turn inhibits edema fluid clearance, due in part to the disassembly of the keratin intermediate filament network, a fundamental element of the cellular cytoskeleton, therefore destructing the epithelial barrier [53]. Therefore, a long-term hypoxia aggravates the disease by inducing more alveolar edema, which forms a vicious circle (Table 1).
Impaired alveolar surfactant production may be another molecular mechanism of alveolar edema in COVID-19 in that the limitation of alveolus superficial active substance would increase alveolar surface tension and hamper alveolar fluid resorption (Table 1) [54][55][56][57][58]. Thus, treatments to alveolar edema may help to both reduce the size of poorly ventilated area and increase the blood oxygen content (Figure 1). Although exogenous surfactant replacements in animal models of ARDS and neonatal respiratory distress syndrome (RDS) showed consistent improvement in gas exchange and survivals, some adult studies have shown only improved oxygenation but no survival benefits [57]. Moreover, a few exogenous pulmonary surfactants have been currently authorized to treat pulmonary permeability edema in COVID-19 patients [57].
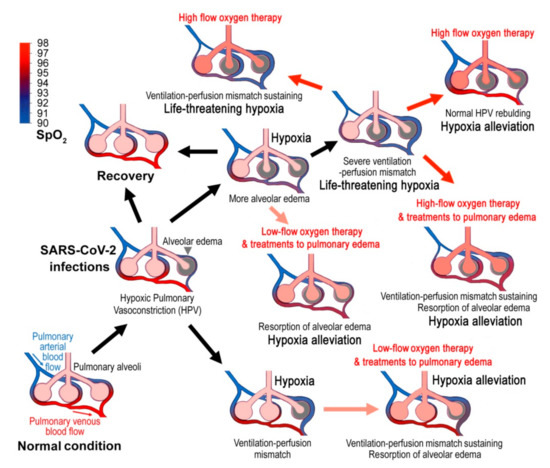
Figure 1. Hypothetically pathogenic mechanisms of COVID-19 and the corresponding therapies. Virus infections cause alveolar edema and the hypoxic pulmonary vasoconstriction (HPV), which is an essential protection mechanism of the lung that directs blood perfusion from badly-ventilated to well-ventilated alveoli to optimize gas exchange. However, SARS-CoV-2 infections may cause mismatched blood flow and alveolar ventilation, and may result in life-threatening hypoxemia. Treatments to pulmonary edema might promote lung edema resorption and show good therapeutic effects. When the disease develops into late stages, systemic alveolar edema and severe ventilation-perfusion mismatch occurs, and the blood oxygen will decline sharply. Then the high-flow oxygen therapy will be required. However, high-flow oxygen inhalation may not correct ventilation-perfusion mismatch and cause several adverse effects. The blue to red gradient bar shows blood oxygen saturation (SpO2).
Besides exogenous surfactant replacements, other treatments that counter-balance the inhibition of edema clearance during hypoxia or improve the lung’s ability to clear alveolar edema should also be adopted. For example, a relatively high partial pressure of O2 in the alveolar gas facilitates alveolar fluid resorption by activating Na+ transport across the alveolar epithelium, which makes an osmotic gradient responsible for the lung edema clearance [53]. Hereby, appropriate oxygen inhalation would accelerate the resorption of pulmonary edema fluid. However, simple oxygen therapy would not achieve adequate effects on alveolar edema clearance. Ethanol/butanol-oxygen vapor therapy (oxygen inhalation with 20% ethanol or butanol as humidifying agent) is a common treatment to pulmonary edema with a long history of safety [60][61][62][63]. Ethanol/butanol vapor decreases the surface tension of the foam inside the pulmonary alveoli and therefore alleviating alveolar edema (Table 2) [60][61][62][63]. Moreover, conservative fluid management, in which diuretics may be administered and intravenous fluid administration is minimized, would reduce hydrostatic pressure and enhance serum oncotic pressure, and therefore limit the development of pulmonary edema potentially (Table 2) [64][65][66]. Unfortunately, these common therapies to pulmonary edema have not attracted enough attention in COVID-19 clinical practice.
Autopsy showed that pulmonary fibrosis was not serious in dead patients with SARS-CoV-2 infections. Intact alveoli could still be seen, but exudation was serious [51][52][67][68][69][70], suggesting the severe alveolar edema. COVID-19 has a prominent feature, that is, a large amount of mucus (edema fluid) could be found in the small airway [51][52][67][68][69][70], which is distinct from other acute pulmonary injuries.
When the disease develops into late stages, systemic alveolar edema and severe ventilation-perfusion mismatch occur, and the blood oxygen will decline sharply. Then the high flow oxygen therapy and mechanical ventilation will be required (Figure 1). However, high-flow oxygen inhalation inhibits HPV because of the reversed diffusion of oxygen, that is, if enough oxygen could bind the receptor in the small alveolar-capillary-arteriole space, the vessels will not vasoconstrict [71]. Upon a high-flow and high-concentration oxygen inhalation, the reflex stimulation to respiration by hypoxia will disappear, resulting in a more serious retention of CO2, which may lead to the hypercapnic encephalopathy (hypercapnic coma) or even a respiratory arrest [22]. Furthermore, high inspired oxygen concentrations could lead to pulmonary-specific toxic effects, such as denitrogenation phenomena, inhibition of surfactant production and severe acidosis, and therefore may worsen ventilation-perfusion mismatch or induce a degree of hypoventilation (Table 2) [72][73]. On the other hand, mechanical ventilation may cause mucus to be blown into the deep of the small airways, which then aggravates intrapulmonary shunt and alveolar hypoxia (Table 2) [74]. These deleterious effects may be an important reason for the high mortality after high-flow oxygen inhalation and mechanical ventilation (The ICU mortality rate among those who required non-invasive ventilation was 79% and among those who required invasive mechanical ventilation was 86%.) [59]. Therefore, nebulized heparin treatments, ethanol-oxygen vapor therapy should be given at the early stages of COVID-19.





