1000/1000
Hot
Most Recent
 +1 point
+1 point
DDR is activated in response to different endogenous and exogenous stresses(). When aberrantly repaired, DNA damage might be associated with clinical outcomes such as neurodegeneration, infertility, and genomic instability, being a key contributing factor to neoplastic transformation and tumour development(). Due to the complexity underneath detection and repair of DNA damage, cells evolved an intricate DDR network that, together with cell cycle regulation, promotes the maintenance of genomic stability and cellular viability().
Cancer is a major burden of disease and one of the leading barriers to improve life expectancy worldwide [1]. Despite many efforts to maximize cancer prevention, diagnosis and treatment, the incidence and mortality rates have been steadily increasing [1][2].
As a highly heterogeneous disease, cancer displays unique genomic and epigenetic variations among patients. As such, the success of conventional chemotherapeutics has been limited by the heterogeneity of patients’ response, development of resistance and severity of side effects [3][4]. Conversely, targeted therapies take advantage of specific alterations in cancer cells, having emerged as a hopeful strategy to overcome these limitations [5][6][7][8].
There is a large amount of evidence that dysfunctional DNA damage repair (DDR) processes are frequently observed in cancer and are associated with genomic instability [5][7][9]. In fact, although cells own an equipped machinery to repair DNA toxic lesions, it may fail, predisposing them to accumulate DNA damage. The survival and proliferation of unrepaired DNA defective cells lead to the accumulation of mutations and genomic instability, strongly contributing to cancer development [5][10]. Indeed, DDR-deficient cells are frequently associated with hypersensitivity to DNA-damaging agents [11][12][13][14][15].
Breast cancer susceptibility gene 1 (BRCA1) is a tumour suppressor gene extensively involved in maintaining genomic integrity through multiple functions in DDR, transcriptional regulation, cell cycle checkpoint and protein ubiquitination [16][17][18]. BRCA1 is frequently dysfunctional in human breast, ovarian, pancreatic, among other cancers, contributing to the accumulation of genomic defects. Despite the increased risk conferred byBRCA1mutations to cancer onset, pre-clinical and clinical data have ascertained that BRCA1 impairment is commonly associated with chemosensitivity in cancer cells [13][16]. Other players involved in DDR can also be found defective in cancer, including breast cancer susceptibility gene 2 (BRCA2),RAD51,RAD52, partner and localizer of BRCA2 (PALB2), ataxia-telangiectasia mutated (ATM) and ataxia telangiectasia and
This review aims to emphasize the potentiality of the DDR pathways, particularly of BRCA1 and interconnected molecules, in precision cancer therapy.
DDR is activated in response to different endogenous and exogenous stresses [5][19] (Figure 1). When aberrantly repaired, DNA damage might be associated with clinical outcomes such as neurodegeneration, infertility, and genomic instability, being a key contributing factor to neoplastic transformation and tumour development [8] Due to the complexity underneath detection and repair of DNA damage, cells evolved an intricate DDR network that, together with cell cycle regulation, promotes the maintenance of genomic stability and cellular viability [20]
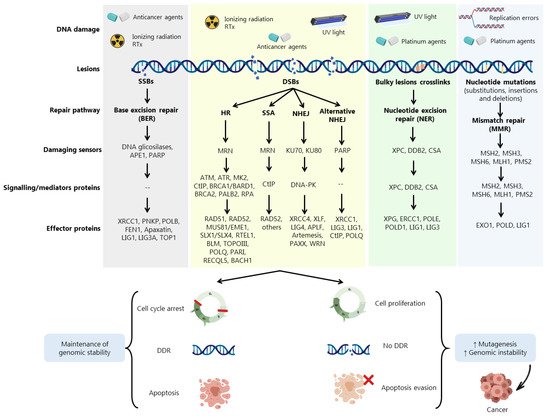
Cells harbouring defects on a particular DDR pathway may compensate by becoming reliant on another repair pathway. In fact, despite showing partially overlapping functions, DDR pathways still exhibit different functionalities depending on multiple damage sensors, signalling factors (activators of cell cycle checkpoints) and effector DDR proteins Conversely, misrepaired DSBs may generate mutations or chromosome rearrangements that may lead to a malignant condition [21]. Three main mechanisms are required for DSBs repair: (i) damage detection, (ii) ability to control cell cycle and transcriptional programs, and (iii) mechanisms for catalysing the repair of the lesion [21].
Accumulated data have shown the involvement of DDR proteins in different stages of cancer development. Early stages of tumorigenesis have been associated with activation of DDR proteins due to the induction of replication stress and DNA damage, acting as a barrier to the proliferation of aberrant cells [20]. However, most of pre-malignant cells are able to escape this barrier by loss or aberrations in specific proteins associated with DDR and cell cycle regulation, such as BRCA1, BRCA2, ATM, RAD51, Fanconi anemia group A protein (FANCA) and p53, allowing these cells to evolve to malignant carcinomas (Figure 1). In more advanced stages, when the tumour is already established, (re)activation and overexpression of DDR factors support cells to evade the lethal effect of the therapeutic agents, eliciting resistance [20].
In 1990,BRCA1(located on chromosome 17) was identified as a classical tumour suppressor gene (TSG) due to the loss of a wild-type (wt) allele during tumorigenesis, being the first TSG associated with hereditary and sporadic cases of basal-like breast cancer [22][23].
Despite being a multifunctional protein, the BRCA1 tumour suppressive function is mainly ensured by its ability to maintain genomic integrity through regulation of diverse cellular processes, including DDR, cell cycle checkpoint, apoptosis, chromosome instability, among others [16][17]. Upon DNA damage, the opposite roles played by p53-binding protein 1 (53BP1) and BRCA1 seem to support cells in the switch between NHEJ and HR [24]. In fact, DNA repair pathways compete to select which mechanism should be employed. This choice is based on several factors, including cell cycle phase.
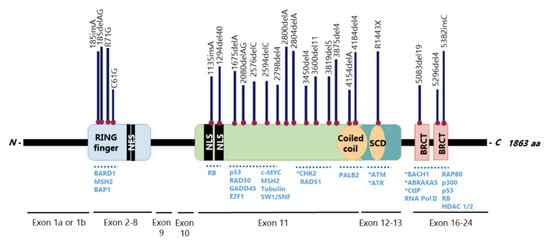
TheBRCA1gene has 24 exons, two of them untranslated, and encodes a large 1863-amino acid phosphoprotein that harbours multiple functional domains, including the highly conservedN-terminal zinc-finger Over 60% of theBRCA1gene is composed by a centrally located exon 11, which encodes two NLS and binding sites for several proteins [25][26] (Figure 2). This is one of the largest human exons (encoding 1142 amino acids) that partially contributes to BRCA1 nuclear localization and activity on cell cycle regulation and DNA repair, being highly required for a functional HR [27][28]. Together with exons 12 and 13, exon 11 encodes a coiled-coil domain that mediates interactions with PALB2 and a serine cluster domain (SCD) that is phosphorylated by ATM and ATR [25][26].
The BRCA1N-terminal RING domain dimerizes with BRCA1-associated RING domain (BARD1), forming stable heterodimers that enhance E3 ubiquitin-ligase activity and DDR [29][30][31][32][33][34] In addition, BRCA1 can undergo proteolytic degradation upon disruption of the BRCA1-BARD1 heterodimer [35][36] (Figure 3). The specific function of the BRCA1-BARD1 heterodimer, its dissociation and interaction with other proteins might be regulated by post-translational modifications as phosphorylation and ubiquitination [30] (Figure 3). Thus, the BRCA1-BARD1 heterodimer plays a crucial role in tumour suppression, interacting with proteins involved in cell cycle, DNA repair, chromosome stability, chromatin modulation, replication fork stability, transcription, among others [37][38]
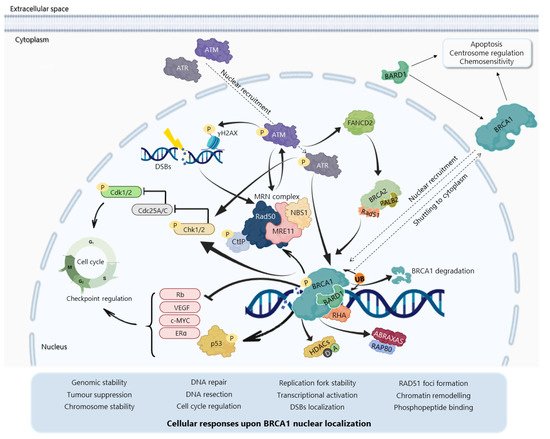
The BRCA1 BRCT domain functions as phosphopeptide recognition modules that enables the BRCA1 binding to phosphorylated partners as BRCA1 Upon DNA damage, BRCA1 phosphorylation by ATM and ATR leads to post-translational modifications of BRCA1-binding proteins and to the subsequent activation of several associated proteins, including checkpoint kinases 1/2 (Chk1/2) and p53, which regulate cell cycle checkpoints [39] BRCA1 also regulates chromatin structure through acetylation and deacetylation of histone proteins by interaction with multiple histone deacetylases (HDAC1 and HDAC2) Thus, due to its rich functional domains, BRCA1 interacts with several transcriptional factors and numerous proteins encoded by tumour suppressors, oncogenes, DNA repair genes, cell cycle regulators, ubiquitin hydrolases and ligases, signalling transducers and chromatin modifying proteins (Figure 2), supporting the complex network involving BRCA1
In DSBs repair by HR, BRCA1 participates as a central component of macromolecular protein complexes, each one composed of unique protein binding partners, as phosphorylated ABRAXAS, BACH1 and CtIP [8][24][40][41] (Figure 4). These complexes, called BRCA1 A, B, C and D helped to recognize the multiple functions of BRCA1 not only in DDR, but also in the transcriptional regulation of genes involved in other cellular processes [40][42]. (Figure 4), with distinct and sometimes overlapping roles for maintenance of genomic stability [40].
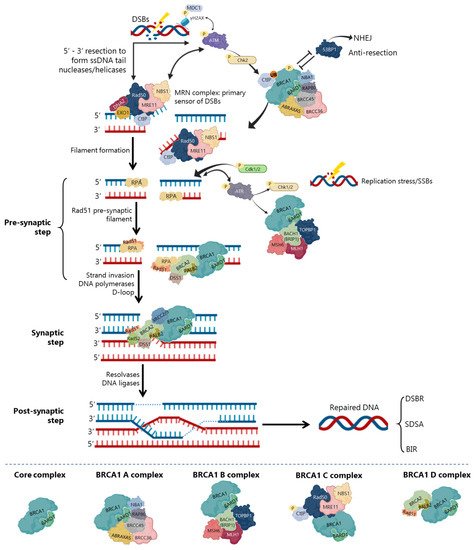
The repair of replication forks or DSBs is initiated by DNA strand resection to generate a 3′-tailed single stranded DNA (ssDNA) In HR, the MRN complex forms a physical bridge, spanning the DSBs end to recruit/retain ATM at DSBs sites. ATM phosphorylates histone H2AX (γH2AX), which recruits the mediator of DNA damage checkpoint 1 (MDC1), enhancing ATM phosphorylation and promoting recruitment of MRN and BRCA1 In the initial phase of HR, CtIP physically interacts with MRN complex (Figure 4), facilitating 5′ end-resection of DSBs.
Precluding the formation of secondary structures at the ssDNA, RPA occupies the 3′-tailed ssDNA derived from DNA end resection, protecting DNA tail from nucleolytic attack and removing the secondary structure [43]. To exchange RPA and facilitate RAD51 loading onto DNA, some mediators, like BRCA2 and PALB2, participate in RAD51-mediated pre-synaptic filament formation, a key intermediate that catalyses homologous pairing and initiates DNA strand invasion [44][26][37][43]. The BRCA1-BARD1 heterodimer helps RAD51-coated ssDNA to invade double stranded DNA (dsDNA) with homologous sequences, enhancing its ability to form a displacement loop structure (D-loop) [24][37]. Accordingly, BARD1 mutations or deletions at residues 758–1,064 of BRCA1 (harbouring the RAD51-interaction domain) abolish the BRCA1-BARD1 ability to promote D-loop and synaptic complex formation [26][37], compromising HR activity and RAD51 nuclear localization [30][45].
Finally, with the support of DNA polymerases, the second end of the damaged chromosome is captured, and anneals to the complementary strand of the intact homologous DNA template [44][24]. In the DSBs repair (DSBR), after priming DNA synthesis and sealing the break, the second end is captured, and a double Holliday junction intermediate is formed. After DNA synthesis and strands ligation, the two Holliday junctions can be resolved by the catalytic function of resolvases to generate crossover products, or dissolved to generate non-crossover products and complete the repair [44][46]. In the synthesis-dependent strand annealing (SDSA) model, the invading strand is displaced from a D-loop by helicase activity and annealed with the 3’ single-stranded tail to complete DNA synthesis and repair.
BRCA1Mutcarriers are at high risk for developing different types of cancer, including breast, ovarian, pancreatic, prostate, laryngeal and fallopian tube cancers [25][47][48]. Since its discovery, more than 1600 mutations have been identified inBRCA1[49], such as frameshift insertions/deletions, nonsense truncation mutations that lead to premature chain termination, and many single nucleotide polymorphisms in the coding or noncoding sequences. Also, a number of missense BRCA1 mutations present clinical relevance, being associated with increased risk of both hereditary and sporadic cancers [47][50]. Heterozygous BRCA1Mutare commonly related to genetic deficiencies in other TSGs and DDR factors, such as phosphatase and tensin homolog (PTEN),ATM/ATR,CHEK2andTP53[16][42][47].
Some highly prevalent pathogenic BRCA1 mutations are more frequent in isolated groups (founder mutations), supporting the existence of distinct incidences among the world population. Although initially described with complete loss of BRCA1 expression, BRCA1Mut:185delAG alleles escape degradation, being translated from an alternative site downstream of the stop codon, which results in a RING-less protein [51]. Besides the loss of BRCA1-BARD1 interaction and subsequent E3 ligase activity, this alternative translation produces a stable and HR-proficient protein that retains the capability to interact with DNA and HR proteins [42][51]. Conversely, BRCA1Mut:185delAG presents a remarkable decrease in BARD1 binding ability [18].
Mutations at BRCT domains commonly present loss of protein expression associated with reduced transactivation activity, growth suppression [52] and aberrant cellular localization [53]. However, recent studies have shown that the truncated mRNA seems to encode a stable protein with potentially new cellular functions due to distinct protein-protein interactions [53]. homozygous BRCA1Mut:5382insC are associated with deficient HR activity and chemosensitivity [54]. Indeed, although BRCA1Mutat BRCT domain has been described as associated with chemosensitivity, the breast HCC1937 cancer cells displayed resistance to PARPi related to residual HR activity by retaining the integrity of RAD51 binding region [55].
Besides somatic mutations,BRCA1promoter hypermethylation and decreasedBRCA1expression, by epigenetic silencing or gene depletion, can also render a dysfunctional BRCA1 pathway in non-hereditary cancers [16][56]. These mechanisms are described to likely contribute to the “BRCAness genotype”, associated with similar biological and clinical phenotypes to that of tumours harbouring BRCA1Mut. However, whether BRCAness mechanisms confer the same functional deficiency is still unclear [56].
A strong connection between triple-negative breast cancer (TNBC) In fact, over 80% of BRCA1Mutbreast cancers are TNBC [57], which is an aggressive form of the disease. Although initially responsive to chemotherapy, most TNBC patients quickly relapse and acquire therapeutic resistance [58][59]. TNBC with pathogenic BRCA1Mutalso demonstrates to be particularly sensitive to platinum and PARPi agents, in both neoadjuvant and adjuvant settings [59].
Despite the lower prevalence, ovarian cancer is three-fold more deadly than breast cancer, with over 70% of patients having late-stage disease [60][61]. In fact, over 10–15% of ovarian cancers are related to germlineBRCA1andBRCA2mutations. Also, these tumours have a dysfunctional DDR pathway that initially promotes sensitivity of cancer cells to chemo- and radiotherapy, although the cell population ultimately ends up developing therapeutic resistance [8]. Indeed, most BRCA1Mut-related ovarian cancer patients experience relapse associated with platinum resistance [62].





