 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | ELENA OBRADOR | + 1924 word(s) | 1924 | 2021-07-23 05:51:27 | | | |
| 2 | Catherine Yang | Meta information modification | 1924 | 2021-07-26 03:24:27 | | |
Video Upload Options
Amyotrophic lateral sclerosis (ALS) is the most common neurodegenerative disease of the motor system. It is characterized by the degeneration of both upper and lower motor neurons, which leads to muscle weakness and paralysis. ALS is incurable and has a bleak prognosis, with median survival of 3–5 years after the initial symptomatology.
1. Introduction
ALS is a rapidly progressive neurodegenerative disease [1] characterized by a gradual loss of upper and lower motor neurons (MNs). The consequence of the loss of MNs in the cerebral cortex, brainstem and medulla is a muscular weakness that progresses, threatening motor autonomy, oral communication, swallowing and/or breathing. The disease does not affect eye muscles, sphincter control or skin sensitivity, nor does it usually cause damage to the patient’s cognitive ability, although approximately 10–15% of cases may present signs of frontotemporal dementia, and in more than 35% of cases, signs of cognitive impairment are detected [2].
The most common form of ALS is called sporadic ALS (SALS) and can affect anyone regardless of gender or ethnicity, although it most often affects people between the ages of 40 and 70. The other type of ALS (5 to 10%) is called familial ALS (FALS), when there is a previously diagnosed first- or second-degree family history [1]. The onset will be spinal in the form of distal weakness in the hands or lower limbs. The slow initial progression and the absence of pain cause patient delay in seeking medical advice. The appearance of abnormal muscle movements (spasms, jerks, cramps or weakness) in the hands and feet causes difficulty in lifting, walking or using one’s hands to dress, wash and button clothing. It can also start with an abnormal loss of muscle mass or body weight [2][3]. The progression of the disease is normally asymmetric. Sometimes, it is very slow, developing over the years and having periods of stability with a variable degree of disability. Other times, its development is rapid [3]. More striking is the bulbar onset, with impaired speech or swallowing. Approximately 3% of cases will make a respiratory debut. Gordon et al. [4] also proposed a diagnostic classification of upper MN-predominant MN disease that mainly distinguishes between clinically pure primary lateral sclerosis and a new category named upper MN-dominant ALS, where patients show spastic paralysis, hyperreflexia, minor electromyogram denervation or lower MN signs on examination and a slower disease progression compared to those with typical ALS [4]. Therefore, ALS should be considered a clinical syndrome, with different forms of presentation and, likely, with multiple risk factors. Regardless of the type of presentation, the ALS patient becomes totally dependent until death, generally due to respiratory failure [5].
Since the vast majority of patients do not have the inherited type of ALS, the diagnosis of ALS is not determined by genetic testing. Unfortunately, there is no specific test that provides a diagnosis, meaning it is currently based on clinical symptoms, the presence in the physical examination of signs of first and second MNs and the electromyographic examination according to criteria from the El Escorial World Federation of Neurology ( http://wfnals.org accessed on 5 June 2021). The rest of the clinical examination and tests will be conducted to exclude diseases that can present similar symptoms and are potentially treatable [6].
The majority of patients do not have a family history of ALS [7][8]. Although more than 100 potentially causative or disease-modifying genes have been identified, the most common mutations are found in C9orf72 (chromosome 9 open reading frame 72), SOD1 (superoxide dismutase 1), FUS (fused in sarcoma/translocated in liposarcoma or heterogeneous nuclear ribonucleoprotein P2) and TARDBP (transactive response DNA binding protein 43) (60–70% of all FALS cases) [9]. Inheritance, in most cases, is autosomal dominant, although autosomal recessive and X-linked dominant familial ALS can also occur [10]. Nevertheless, not all family members with the same mutations necessarily develop FALS. On the other hand, in the dark well of possible risk factors (causes) of SALS, there is a large list of suggested factors, i.e., (but not limited to) neurotoxins, chemicals, metals, radiation and electromagnetic fields, sport-related stress, non-equilibrated dietary habits, infectious agents, smoking, autoimmunity or traumatic brain injuries [11]. Most or all of them could be just contributing or enhancing factors in the complex pathophysiology that leads to the death of MNs. Some of the pathological alterations could even begin before the onset of motor symptoms. Whatever the real course of the pathophysiology, many of the abovementioned factors directly or indirectly converge in the mitochondria.
2. Glia, Neuroinflammation and Motor Neuron Mitochondria
Neuroinflammation has been consistently observed in motor regions of the central nervous system (CNS) in SALS and FALS, constituting a hallmark of the disease [12].
Microglia cells represent the first and primary form of active immune defense in the CNS and can be activated by any type of toxic challenge, including injury, infection and ischemia [13]. Upon activation, they enlarge, migrate and can become phagocytic to remove toxic matter. Activated microglial cells secrete pro-inflammatory and neurotoxic factors [14]. If the microglia cells cannot resolve or eliminate a toxic insult in the CNS, they remain reactive and continue to recruit astrocytes and oligodendrocytes, causing an ongoing inflammatory process [15]. In rodents expressing the ALS-linked SOD1 G93A mutation (a G93A transgene mutant form of human SOD1), rapid spread of paralysis coincides with the emergence of neurotoxic and proliferating aberrant glia cells with an astrocyte-like phenotype (found surrounding the damaged MNs) [16]. However, selective NF-κB inhibition in ALS astrocytes was not found sufficient to rescue motor neuron death [17]. However, the deletion of NF-κB signaling in microglia rescued MNs from microglial-mediated death in vitro and extended survival in ALS mice [17], thus suggesting anti-inflammation and inhibition of microglia-induced toxicity as a potential therapeutic strategy.
Furthermore, in rat astroglial cultures exposed to the cerebrospinal fluid from ALS patients, enhanced production and release of inflammatory cytokines IL6 (interleukin 6) and TNFα, as well as increased production of cyclooxygenase-2 (COX-2) and prostaglandin E2 (PGE2), have been observed. Concomitantly, anti-inflammatory cytokine IL10 and the beneficial trophic factors VEGF (vascular endothelial growth factor) and GDNF (glial cell-derived neurotrophic factor) were down-regulated [18]. Moreover, in experiments where reactive/aberrant astrocytes were co-cultured with MNs, neuron survival was less than 10% [19], thus suggesting a non-permissive microenvironment for neuron growth and differentiation. Human astrocytes derived from the spinal cord of persons with SALS or FALS also killed MNs in culture [20].
There is evidence linking mitochondrial dysfunction and neuroinflammation [21][22][23]. Different studies have found a link between TANK-binding kinase 1 (TBK1) mutations and ALS. TBK1 is an inducer of type-1 interferons and also has a key role in autophagy and mitophagy [24]. Fragmented mitochondria also fuel neuroinflammation [25]. For instance, mitochondria fission was found to be mediated by the Drp1-Fis1 signaling pathway, which was blocked by P110 [25], thus suggesting that targeted inhibition of mitochondria fission may represent a new strategy for preventing nerve cell death in ALS. Moreover, in a recent contribution where the effect of an NAD + booster (nicotinamide riboside) and a natural antioxidant (pterostilbene) was assayed in ALS SOD1 G93A mice, the interdependence among neuroinflammation, mitochondria and the activation of apoptotic cell death in MNs was clearly described [26]. Importantly, nicotinamide riboside and pterostilbene also decreased the microgliosis and astrogliosis associated with ALS progression [26].
3. Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Mechanisms Leading to the Death of Motor Neurons
Oxidative stress, capable of damaging all components of a cell, is the consequence of an increased generation of ROS and/or a decrease in the effectiveness of antioxidant defenses [27]. Although ROS may not be the triggering factor for neurodegenerative diseases (including ALS), they are likely to exacerbate disease progression [12]. Resident glial cells and infiltrated immune cells are considered among the major producers of ROS and reactive nitrogen species (RNS) in pathological conditions of the CNS, including MN diseases [28]. NO and H 2O 2 produced in excess can lead to the formation of potent oxidants, such as .OH and - OONO radicals, via a trace metal-dependent process [29].
Besides MNs, mitochondria, as it occurs in any cell, are the primary site of energy generation, but also the main source of ROS. Under conditions in which mitochondrial generation of ROS is increased (such as in the presence of Ca 2+ ions or when the mitochondrial antioxidant defense mechanisms are compromised), these reactive species may lead to irreversible damage of mitochondrial DNA, membrane lipids and proteins. Oxidative stress-induced damage can impair mitochondrial function and ultimately cause cell death [30].
The redox state is often used to describe the balance of glutathione disulfide/glutathione (GSSG/GSH), NAD + /NADH and NADP + /NADPH in cells and is reflected in the balance of several sets of metabolites (e.g., lactate and pyruvate, β-hydroxybutyrate and acetoacetate), whose interconversion is dependent on these ratios (e.g. , Jones 2015 [31]). For instance, GSH levels (the main non-protein thiol and a prevalent antioxidant in mammalian cells) are lower in the motor cortex of ALS patients as compared to healthy volunteers [32]. It has been shown that GSH depletion promotes neurological deficit, mitochondrial pathology [33] and MN degeneration [34] in mutant SOD1 ALS mice. Regarding toxic protein aggregates, Meyerowitz et al. observed that chronic oxidative and nitrosative stress induced several features consistent with TDP-43 (the polypeptide product of the TARDBP gene) proteinopathies including loss of nuclear TDP-43, accumulation of diffuse TDP-43 in the cytosol, formation of a 35-kDa C-terminal fragment and accumulation of TDP-43 in RNA stress granules, some of which revealed ubiquitination [35]. Both sporadic and familial forms of ALS have the pathologic TDP-43 signature of abnormal hyperphosphorylation, ubiquitination and C-terminal fragments in affected brains and spinal cords, suggesting that they share a common mechanism of pathogenesis. Importantly, it has also been observed that TDP-43 deposition leads to targeted RNA instability in ALS and may ultimately cause cell death by disrupting energy production and protein synthesis pathways [36].
Based on the experimental facts discussed thus far, and the abundant available literature on mitochondrial alterations in ALS, it is feasible to deduce that oxidative stress plays a central role in the pathophysiology of ALS. Figure 1 briefly describes how oxidative stress triggers mitochondrial dysfunctions and the cascade of events that leads to MN degeneration and death.
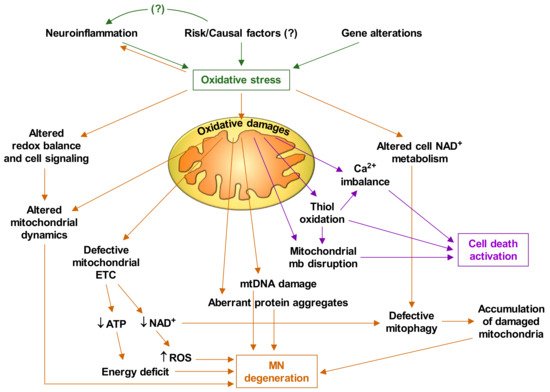
4. Targeting Mitochondria as a Therapy
The association of nicotinamide riboside and pterostilbene (see above) showed efficacy in ALS patients in a small pilot study (NCT03489200 EH301 for the Treatment of ALS) and is now being tested in a larger trial (NCT04562831 the NO-ALS Study).
Metal chelators, e.g., the iron chelator deferiprone, may also help to limit metal-induced toxicity leading to oxidative stress [38]. A clinical trial showed that the decreases in the ALSFRS scale and the body mass index were significantly smaller for the first 3 months of deferiprone treatment as compared to placebo-treated patients (FAIR-ALS II NCT03293069).
Additionally, targeting the clearance of damaged mitochondria is a therapeutic strategy that could be implemented. In this regard, some mitophagy inducers have promising benefits to the protection of neurons, e.g., NAD + precursors (such as nicotinamide riboside), urolithin A [39], spermidine [40] or the FDA-approved rapamycin [41] and metformin [42].
Olesoxime (TRO19622), a small cholesterol-like structure, has shown neuroprotective properties for MNs in cell culture and in rodents acting on mitochondria, possibly at the PTP [43]. However, only in the first 12 months of a phase II/III clinical trial did it show an improvement in the motor symptoms of spinal muscular atrophy (MITOTARGET NCT00868166).
References
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172.
- Brent, J.R.; Franz, C.K.; Coleman, J.M.; Ajroud-Driss, S. ALS: Management problems. Neurol. Clin. 2020, 38, 565–575.
- Takei, K.; Tsuda, K.; Takahashi, F.; Hirai, M.; Palumbo, J. An assessment of treatment guidelines, clinical practices, demographics, and progression of disease among patients with amyotrophic lateral sclerosis in Japan, the United States, and Europe. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 88–97.
- Gordon, P.H.; Cheng, B.; Katz, I.B.; Pinto, M.; Hays, A.P.; Mitsumoto, H.; Rowland, L.P. The natural history of primary lateral sclerosis. Neurology 2006, 66, 647–653.
- Adler, D.; Poncet, A.; Iancu Ferfoglia, R.; Truffert, A.; Janssens, J.-P. Predicting respiratory failure in amyotrophic lateral sclerosis: Still a long way to go. Eur. Respir. J. 2019, 54.
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929.
- Al-Chalabi, A.; Hardiman, O.; Kiernan, M.C.; Chiò, A.; Rix-Brooks, B.; van den Berg, L.H. Amyotrophic lateral sclerosis: Moving towards a new classification system. Lancet Neurol. 2016, 15, 1182–1194.
- Riancho, J.; Gil-Bea, F.J.; Santurtun, A.; López de Munaín, A. Amyotrophic lateral sclerosis: A complex syndrome that needs an integrated research approach. Neural Regen. Res. 2019, 14, 193–196.
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310.
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258.
- Martin, S.; Al Khleifat, A.; Al-Chalabi, A. What causes amyotrophic lateral sclerosis? F1000Research 2017, 6, 371.
- Liu, J.; Wang, F. Role of neuroinflammation in amyotrophic lateral sclerosis: Cellular mechanisms and therapeutic implications. Front. Immunol. 2017, 8, 1005.
- Filiano, A.J.; Gadani, S.P.; Kipnis, J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 2015, 1617, 18–27.
- Dahlke, C.; Saberi, D.; Ott, B.; Brand-Saberi, B.; Schmitt-John, T.; Theiss, C. Inflammation and neuronal death in the motor cortex of the wobbler mouse, an ALS animal model. J. Neuroinflamm. 2015, 12, 215.
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783.
- Jiménez-Riani, M.; Díaz-Amarilla, P.; Isasi, E.; Casanova, G.; Barbeito, L.; Olivera-Bravo, S. Ultrastructural features of aberrant glial cells isolated from the spinal cord of paralytic rats expressing the amyotrophic lateral sclerosis-linked SOD1G93A mutation. Cell Tissue Res. 2017, 370, 391–401.
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-ΚB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023.
- Mishra, P.-S.; Dhull, D.K.; Nalini, A.; Vijayalakshmi, K.; Sathyaprabha, T.N.; Alladi, P.A.; Raju, T.R. Astroglia acquires a toxic neuroinflammatory role in response to the cerebrospinal fluid from amyotrophic lateral sclerosis patients. J. Neuroinflamm. 2016, 13, 212.
- Díaz-Amarilla, P.; Olivera-Bravo, S.; Trias, E.; Cragnolini, A.; Martínez-Palma, L.; Cassina, P.; Beckman, J.; Barbeito, L. Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 18126–18131.
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828.
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative stress, neuroinflammation and mitochondria in the pathophysiology of amyotrophic lateral sclerosis. Antioxidants 2020, 9, 901.
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimer Dis. 2010, 20 (Suppl. S2), S369–S379.
- Wilkins, H.M.; Swerdlow, R.H. Relationships between mitochondria and neuroinflammation: Implications for Alzheimer’s disease. Curr. Top. Med. Chem. 2016, 16, 849–857.
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5.
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W.; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648.
- Obrador, E.; Salvador, R.; Marchio, P.; López-Blanch, R.; Jihad-Jebbar, A.; Rivera, P.; Vallés, S.L.; Banacloche, S.; Alcácer, J.; Colomer, N. Nicotinamide riboside and pterostilbene cooperatively delay motor neuron failure in ALS SOD1 G93A mice. Mol. Neurobiol. 2020, 58, 1345–1371.
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183.
- D’Ambrosi, N.; Cozzolino, M.; Carrì, M.T. Neuroinflammation in amyotrophic lateral sclerosis: Role of redox (Dys) regulation. Antioxid. Redox Signal. 2017, 29, 15–36.
- Carretero, J.; Obrador, E.; Esteve, J.M.; Ortega, A.; Pellicer, J.A.; Sempere, F.V.; Estrela, J.M. Tumoricidal activity of endothelial cells. Inhibition of endothelial nitric oxide production abrogates tumor cytotoxicity induced by hepatic sinusoidal endothelium in response to B16 melanoma adhesion in vitro. J. Biol. Chem. 2001, 276, 25775–25782.
- Kowaltowski, A.J.; Vercesi, A.E. Mitochondrial damage induced by conditions of oxidative stress. Free Radic. Biol. Med. 1999, 26, 463–471.
- Jones, D.P.; Sies, H. The redox code. Antioxid. Redox Signal. 2015, 23, 734–746.
- Weiduschat, N.; Mao, X.; Hupf, J.; Armstrong, N.; Kang, G.; Lange, D.J.; Mitsumoto, H.; Shungu, D.C. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014, 570, 102–107.
- Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked HSOD1(G93A) mice model. Neurobiol. Dis. 2011, 43, 543–551.
- Killoy, K.M.; Harlan, B.A.; Pehar, M.; Helke, K.L.; Johnson, J.A.; Vargas, M.R. Decreased glutathione levels cause overt motor neuron degeneration in HSOD1WT over-expressing mice. Exp. Neurol. 2018, 302, 129–135.
- Meyerowitz, J.; Parker, S.J.; Vella, L.J.; Ng, D.C.; Price, K.A.; Liddell, J.R.; Caragounis, A.; Li, Q.-X.; Masters, C.L.; Nonaka, T.; et al. C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol. Neurodegener. 2011, 6, 57.
- Tank, E.M.; Figueroa-Romero, C.; Hinder, L.M.; Bedi, K.; Archbold, H.C.; Li, X.; Weskamp, K.; Safren, N.; Paez-Colasante, X.; Pacut, C.; et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9, 2845.
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933.
- Moreau, C.; Danel, V.; Devedjian, J.C.; Grolez, G.; Timmerman, K.; Laloux, C.; Petrault, M.; Gouel, F.; Jonneaux, A.; Dutheil, M.; et al. Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis? Antioxid. Redox Signal. 2018, 29, 742–748.
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin a is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603.
- Yang, X.; Zhang, M.; Dai, Y.; Sun, Y.; Aman, Y.; Xu, Y.; Yu, P.; Zheng, Y.; Yang, J.; Zhu, X. Spermidine inhibits neurodegeneration and delays aging via the PINK1-PDR1-dependent mitophagy pathway in C. elegans. Aging 2020, 12, 16852–16866.
- Li, Q.; Gao, S.; Kang, Z.; Zhang, M.; Zhao, X.; Zhai, Y.; Huang, J.; Yang, G.-Y.; Sun, W.; Wang, J. Rapamycin enhances mitophagy and attenuates apoptosis after spinal ischemia-reperfusion injury. Front. NeuroSci. 2018, 12, 865.
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 2020, 32, 44–55.e6.
- Martin, L.J. Olesoxime, a cholesterol-like neuroprotectant for the potential treatment of amyotrophic lateral sclerosis. IDrugs 2010, 13, 568–580.


