The chemokines CCL5 and CXCL4 are deposited by platelets onto endothelial cells, inducing monocyte arrest. Here, the fate of CCL5 and CXCL4 after endothelial deposition was investigated. Human umbilical vein endothelial cells (HUVECs) and EA.hy926 cells were incubated with CCL5 or CXCL4 for up to 120 min, and chemokine uptake was analyzed by microscopy and by ELISA. Intracellular calcium signaling was visualized upon chemokine treatment, and monocyte arrest was evaluated under laminar flow. Whereas CXCL4 remained partly on the cell surface, all of the CCL5 was internalized into endothelial cells. Endocytosis of CCL5 and CXCL4 was shown as a rapid and active process that primarily depended on dynamin, clathrin, and G protein-coupled receptors (GPCRs), but not on surface proteoglycans. Intracellular calcium signals were increased after chemokine treatment. Confocal microscopy and ELISA measurements in cell organelle fractions indicated that both chemokines accumulated in the nucleus. Internalization did not affect leukocyte arrest, as pretreatment of chemokines and subsequent washing did not alter monocyte adhesion to endothelial cells. Endothelial cells rapidly and actively internalize CCL5 and CXCL4 by clathrin and dynamin-dependent endocytosis, where the chemokines appear to be directed to the nucleus. These findings expand our knowledge of how chemokines attract leukocytes to sites of inflammation.
1. Introduction[1]
Chemokines are small chemotactic cytokines that have an important role in regulating leukocyte trafficking during health and disease
[1][2]. Through binding and activation of their cognate G protein-coupled receptors, they can rapidly induce leukocyte responses e.g., integrin activation, flow-resistant arrest, cell polarization, and transendothelial migration to sites of inflammation or infection. On a structural level, chemokines are hallmarked by a disordered N-terminus, a 3-strand antiparallel β-sheet, and a C-terminal α-helix. In addition, stretches of basic amino acids mediate binding to glycosaminoglycans (GAGs), e.g., heparin, heparan sulfate, and similar sulfated polysaccharides that constitute the cellular glycocalyx
[3][4]. This warrants immobilization of the chemokines to the cell surface, e.g., of endothelial cells (EC) of the vessel wall, allowing them to be visible by rolling leukocytes. Besides this concept of chemokine presentation on the endothelial surface, constituting a message for leukocytes, some chemokines might be produced by the EC themselves and stored in small vesicles below the apical cell surface, which can be located by adherent monocytes prior to diapedesis
[5]. In addition, chemokines on the vessel wall might originate from the subendothelial tissue and move to the vascular surface by transcytosis
[6][7], yet they can also be deposited on the vessel wall by rolling platelets, as was shown for CCL5 (RANTES)
[8]. This chemokine transfer to EC by activated platelets was shown to facilitate subsequent monocyte arrest
[8][9]. Infusion of activated platelets into hyperlipidemic mice resulted in an accelerated development of atherosclerosis, which could be attributed in part to increased immobilization of CCL5 onto the atherosclerotic vessels
[10]. Interestingly, CCL5 and CXCL4 (platelet factor 4), one of the most abundant chemokines in platelets, can interact with each other to form heterodimers, which are particularly potent in the recruitment of monocytes
[11] and were shown to modulate the severity of atherosclerosis, stroke, abdominal aneurysm, and myocardial infarction in mice
[12][13][14][15][16]. Although the interaction of CCL5 with GAGs has been postulated as essential for function in vitro and in vivo
[17], the exact mechanism of CCL5 presentation to the cell surface and recognition by immune cells is incompletely characterized. Although the presence of CXCL4 led to increased binding of CCL5 to the surface of monocytic cells, it is unclear whether this explains the synergy between those chemokines
[11]. A previous study has indicated that CCL5 is immobilized on the surface of human umbilical vein endothelial cells (HUVEC) in filamentous flow-resistant polymers, which might form a scaffold for leukocyte recruitment
[18]. Interestingly, part of the CCL5 was observed intracellularly. To elaborate on the previous findings and to further investigate the mechanisms that underlie chemokine-induced leukocyte recruitment, we investigated the fate of exogenously added CCL5 and CXCL4 to EC. We found that incubation of EC with CCL5 and CXCL4 under static conditions led to rapid internalization of the chemokines, where CXCL4 remained partly presented on the cell surface. Internalization was an active process and dependent on G protein-coupled receptor (GPCR) signaling and classic endocytosis and resulted in calcium signaling within endothelial cells. Remarkably, internalized CCL5 and CXCL4 were targeted to the nucleus. Leukocyte arrest was not altered upon pretreatment with chemokines.
2. Rapid Internalization and Nuclear Translocation of CCL5 and CXCL4 in Endothelial Cells
2.1. Surface Presentation of the Chemokines CCL5 and CXCL4 on EC
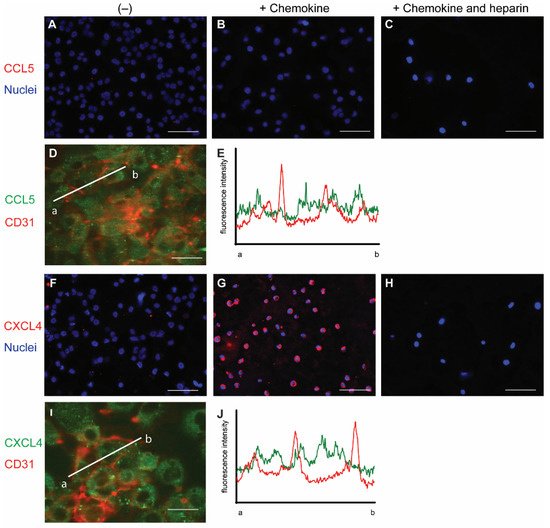
To initially investigate the interaction of chemokines with endothelial cells, cells from the line EA.hy926 (EAHy) were incubated without or with CCL5 and CXCL4, for a prolonged time of 60 min at 37 °C. The cells were subsequently stained using specific fluorescent antibodies without prior permeabilization. Thus, only the extracellular fraction of CCL5 and CXCL4 would be visible. Absence of exogenous chemokines before staining did not result in a notable fluorescent signal for either CCL5 or CXCL4 (
Figure 1A,F). Likewise, the fluorescent intensity did not notably increase after 60 min treatment of EAHy cells with CCL5 (
Figure 1B). However, incubation of EAHy with CXCL4 led to a robust fluorescent signal (
Figure 1G). Washing the EAHy cells with heparin after incubation with chemokines, but prior to antibody staining, led to loss of fluorescent signal (
Figure 1C,H). Co-staining of confluent EAHy cells with CD31 and CCL5 or CXCL4, respectively, revealed a cytoplasmic staining pattern of the chemokines that was distinct from the typical accumulation of the CD31 signal at the cell-cell contacts (
Figure 1D,E,I,J). Imaging along the
z-axis implied faint CCL5 at the luminal aspect of the EAHy cells and increased staining intensities toward the basolateral side (
Figure S1B).
Figure 1. Staining of chemokines after addition to endothelial cells. EAHy were grown on a cell culture slide, mock-treated (A,F), or incubated with the chemokines CCL5 (B–D,) or CXCL4 (G–I) for 60 min at 37 °C, and cells were washed with PBS alone or PBS with 1 mg/mL heparin (C,H). External chemokines on living cells were then stained with the respective primary antibodies and an Alexa Fluor 647-coupled secondary antibody, and nuclei were visualized with Hoechst 33342. (A–C,F–H). Internalized chemokines were stained after fixation and permeabilization of cells using a FITC-coupled secondary antibody (green), and cell membrane was visualized with APC-coupled CD31 (red), using confocal microscopy (D,I). Intensity profile through adjacent endothelial cells indicated by line a-b in (D,I) respectively, showing cell membrane (red) and CCL5 (E) or CXCL4 (J) resp. (green). Scale bar: 100 µm (A–C,F–H) or 50 µm (D,I); (n = 4).
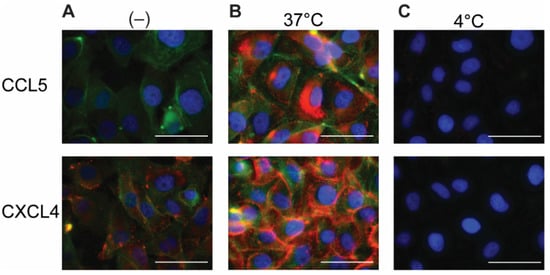
2.2. Permeabilization of EAHy Increases the CCL5 and CXCL4 Antigen Signal
Because chemokines are known to be retained by EC, the EAHy cells were permeabilized in order to investigate an intracellular presence. After permeabilization and addition of the fluorescent antibodies, minimal staining of CCL5 and CXCL4 was observed in the absence of exogenous chemokines (
Figure 2A). This signal might reflect low levels of endogenous CCL5 or CXCL4 (or variant CXCL4L1
[19]) present in EAHy. Interestingly, incubation of EAHy with exogenously added CCL5 and CXCL4 at 37 °C for 60 min, followed by permeabilization and staining, resulted in a high signal intensity of the respective chemokine (
Figure 2B). Incubation of EAHy with chemokines at 4 °C did not lead to an increase in fluorescent signal (
Figure 2C), suggesting that the intracellular accumulation of CCL5 and CXCL4 is an active and energy-requiring cellular process.
Figure 2. Addition of CCL5 and CXCL4 to EC leads to internalization. EAHy incubated with buffer (A) or the chemokines CCL5 (top row) or CXCL4 (bottom row) at 37 or 4 °C (B,C, respectively) for 60 min and washed with heparin (1 mg/mL) prior to fixation, permeabilization, and staining. CCL5 (red, upper row), CXCL4 (red, lower row), F-actin (green), and nuclei (blue). Scale bar: 50 µm. (n = 4).
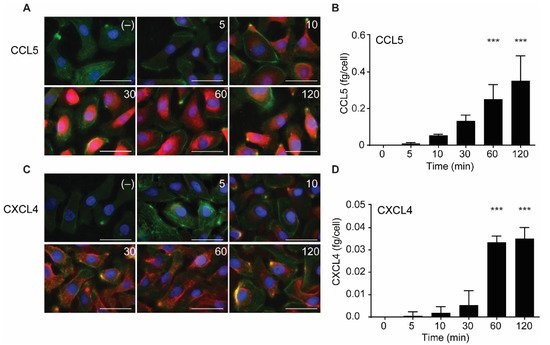
2.3. Intracellular Accumulation of CCL5 and CXCL4 Is Time-Dependent
In further experiments, the uptake of CCL5 and CXCL4 was followed in time. The chemokines were added and remained present at various increasing time points at 37 °C. Then, surface-bound and excess chemokines were removed by washing with heparin, and cells were fixed, permeabilized, and stained with specific fluorescent-labeled antibodies. Subsequently, the presence of the chemokines was visualized using fluorescent microscopy. In addition, the chemokine-treated cells were lysed after washing, and intracellular chemokine concentrations were measured by ELISA.
Both CCL5 and CXCL4 appeared to be taken up in a time-dependent manner (Figure 3). An increase in subcellular fluorescent signal was already observed after 5 min of incubation and increased over the 120-min duration of the experiment (Figure 3A,C). This was paralleled by an increase of intracellular CCL5 and CXCL4 antigen as observed with ELISA, with an apparent maximal uptake of CXCL4 after 60 min (Figure 3B,D). This supported the idea that EAHy cells actively and rapidly take up the chemokines CCL5 and CXCL4. For clarity, images with separate color channels are shown in Figure S1. Bovine chemokines from FCS did not cross-react with either antibodies used for fluorescence or ELISA (data not shown).
Figure 3. Time-dependent uptake of CCL5 and CXCL4 in EC. CCL5 and CXCL4 were added to EAHy for indicated times at 37 °C. After washing with heparin, cells were fixed, permeabilized, and stained. Fluorescent signals of CCL5 (A, red) and CXCL4 (C, red), actin (green), and nuclei (blue) (n = 3). Quantification of intracellular chemokine concentrations of CCL5 (B) and CXCL4 (D) by ELISA. *** p < 0.001 (n = 3), ANOVA with Sidak’s post-test.
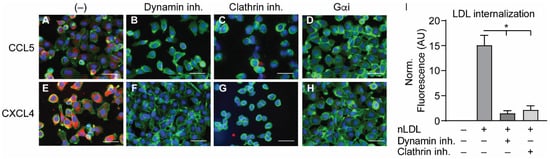
2.4. Internalization of CCL5 and CXCL4 Is Dependent on Dynamin- and Clathrin-Mediated Endocytosis
In order to investigate the manner of chemokine uptake, the EAHy cells were pre-treated with inhibitors of clathrin- (Pitstop2) or dynamin-mediated endocytosis (Dynasore) for 15 min, after which the cells were incubated with the chemokines in the presence of the inhibitors. Then, the same procedure for fixation, staining, and imaging as described above was followed. Of note, the pre-treatment with endocytosis inhibitors led to some detachment of the EAHy cells and loss of monolayer properties. Both inhibitors abolished the intracellular uptake of CCL5 and CXCL4 (
Figure 4A–H). In addition, blockade of chemokine receptor- and other GPCR-induced signaling by pertussis toxin also led to a reduced internal presence of CCL5 and CXCL4. Effective inhibition of clathrin- and dynamin-dependent endocytosis by the above inhibitors was demonstrated by measurement of DiI-labeled nLDL uptake (
Figure 4I). Surprisingly, enzymatic removal of GAGs from the cell surface resulted in an increased uptake of both CCL5 and CXCL4 into EAHy cells (
Figure S1). Although chemokine uptake was abolished by pertussis toxin, antagonists of the CCL5 receptor and of the putative CXCL4 receptor CXCR3 prevented neither CCL5 nor CXCL4 internalization (
Figure S2). Interestingly, pre-incubation of EAHy cells with CXCL4 prior to the addition of CCL5 led to a significant reduction of subsequent CCL5 uptake (
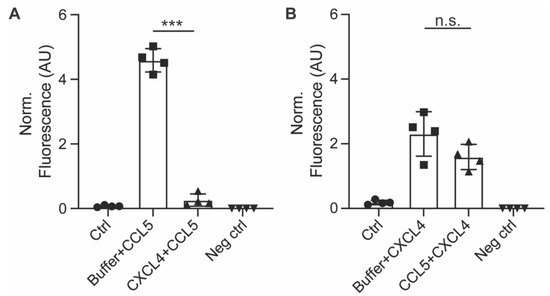
Figure 5A). By contrast, pre-incubation of the cells with CCL5 prior to CXCL4 addition did not affect CXCL4 uptake. This indicates that CXCL4 can desensitize EAHy cells for the uptake of CCL5 (
Figure 5B).
Figure 4. Internalization of CCL5 and CXCL4 is dependent on dynamin- and clathrin-mediated endocytosis, and on G protein-coupled receptor signaling (A–H). EAHy were pre-incubated without (A,E) or with inhibitors to dynamin (B,F), clathrin (C,G), or GPCR (D,H) prior to incubation with CCL5 (top row) or CXCL4 (bottom row) at 37 °C. Cells were then washed with heparin prior to fixation and permeabilization and stained for the chemokines (red), F-actin (green), and nuclei (blue). Scale bar: 100 µm. Panel (I) shows the internalization of LDL in EAHy, and the blocking thereof by the inhibitors to dynamin or clathrin, as a positive control. * p < 0.05, ANOVA with Dunn’s post-hoc test (n = 6).
Figure 5. Pre-incubation with CXCL4 inhibits subsequent uptake of CCL5. EAHy cells were pre-incubated with CXCL4 for 30 min prior to incubation with CCL5 for 120 min and its uptake measured by antibody staining after heparin washing (A), or vice versa (B). *** p < 0.001 (n = 4), ANOVA with Sidak’s post-test. n.s. = non-significant.
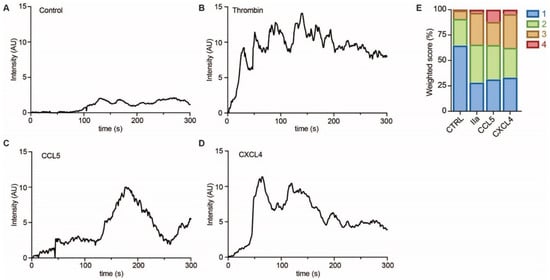
To investigate whether EC showed evidence of intracellular signaling upon treatment with CCL5 or CXCL4, HUVECs were loaded with a calcium-sensitive dye and treated with buffer, thrombin as a positive control, or chemokines (Figure 6A–D). Upon stimulation with CCL5 and CXCL4, the HUVECs showed a notable and lasting increase in intracellular calcium levels, which was comparable to that induced by thrombin, as evidenced by classification of the individual cellular calcium mobilization profiles (Figure 6E).
Figure 6. Chemokines induce calcium signaling in EC. HUVECs loaded with Fluo-4 were stimulated with PBS (control, A), thrombin (IIa, B), CCL5 (C), and CXCL4 (D), and intensity profiles (in arbitrary units, AU) were recorded of >30 cells in over 3 independent experiments. Representative traces are shown in (A–D) and traces were classified as described in the methods section and summarized in (E).
2.5. CCL5 and CXCL4 Are Targeted to the Nucleus after Endothelial Uptake
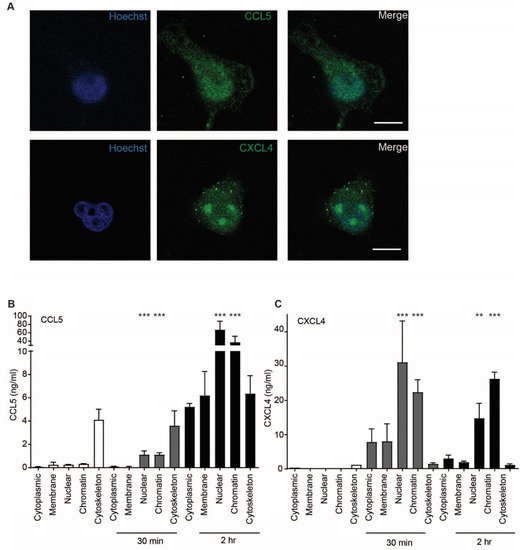
Because uptake of chemokines appeared to be an active process accompanied by cell signaling events, the intracellular fate of CCL5 and CXCL4 was investigated further. Confocal microscopy indicated that the chemokines accumulated in the nucleus 60 min after addition (Figure 7A). This is supported by a high optical resolution Z-stack movie that suggests accumulation of CXCL4 in the nucleoli (Supplementary Movie S1). In order to further elucidate the subcellular localization of CCL5 and CXCL4, EAHy were lysed after 30 and 120 min of treatment. Subcellular fractions were isolated and measured. Interestingly, both CCL5 and CXCL4 showed an association with the cytoskeleton after 30 min, with a slight further increase at 120 min. However, the nuclear content was only barely increased after 30 min, but strongly increased after 120 min (Figure 7B,C). These data suggest that both CCL5 and CXCL4 are transported to the nucleus through cytoskeletal associations. To investigate whether inflammatory conditions could affect the uptake of chemokines, EAHy were activated using TNFa for 4 or 18 h, prior to addition of CCL5 or CXCL4. However, no difference in chemokine uptake was observed compared with resting cells (Figure S3).
Figure 7. CCL5 and CXCL4 are targeted to the nucleus. (A) EAHy were incubated with CCL5 or CXCL4 for 60 min at 37 °C, fixed, and stained for nuclei (blue), chemokine CCL5, and CXCL4 (green). Shown are confocal micrographs. Scalebar = 10 µm. (n = 3) (B,C) EAHy were incubated without (white bars) or with CCL5 (B) or CXCL4 (C) for 30 or 120 min (gray and black bars, respectively), lysed, and chemokines were determined in subcellular fractions (n = 3, ** p < 0.01, *** p < 0.001, ANOVA with Dunn’s post-test).
2.6. CCL5 and CXCL4 Internalization Does Not Affect Leukocyte Arrest
Previous findings suggested that chemokines e.g., CCL2, stored in subluminal vesicles, could guide lymphocyte tracking on EC
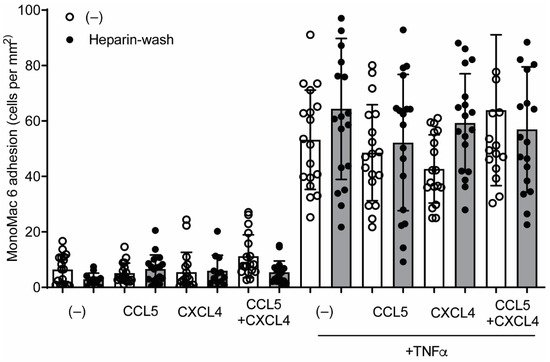
[5]. In order to investigate whether a similar mechanism could regulate CCL5- and CXCL4-induced monocyte arrest, HUVECs were incubated with these chemokines for 120 min, treated without or with heparin to remove residual surface chemokines, and subsequently perfused with monocytic MonoMac6 cells. Addition of chemokines for 120 min did not affect MonoMac6 adhesion to HUVECs (
Figure 8), indicating that internalized chemokines neither make HUVECs competent for leukocyte interactions nor directly induce leukocyte arrest. Similar results were observed after activation of the HUVECs with TNFa for 4 h, prior to the addition of chemokines (
Figure 8). The combined addition of CCL5 and CXCL4, which is a potent stimulus for monocyte arrest, did not increase MonoMac6 adhesion, which indicates that TNFa activation might already support maximal monocyte arrest. These results suggest that at least CCL5 and CXCL4 require surface presentation for leukocyte recruitment.
Figure 8. CCL5 and CXCL4 internalization does not affect monocytic cell arrest to endothelial cells. HUVECs were stimulated with 10 ng/mL TNFa for 4 h and/or CCL5 or CXCL4 for 1 h and washed without (black bars) or with heparin (white bars) prior to perfusion of Syto 13-labeled MonoMac-6 cells at 3 dynes/cm2. Arrested cells were counted in 6 different fields (n = 4).
 +1 point
+1 point