 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christos Damaskos | + 4582 word(s) | 4582 | 2021-07-13 07:54:54 | | | |
| 2 | Amina Yu | Meta information modification | 4582 | 2021-07-20 07:39:16 | | |
Video Upload Options
Triple-negative breast cancer (TNBC) is an aggressive subtype of breast cancer (BC) and accounts for 10–20% of cases. Due to the lack of expression of several receptors, hormone therapy is largely ineffective for treatment purposes. Nevertheless, TNBC often responds very well to chemotherapy, which constitutes the most often recommended treatment.
1. Introduction
Breast cancer (BC) is considered the second most commonly occurring pathology in the world [1]. BC is more frequently diagnosed in less developed and industrialized countries, it also constitutes the second notable cause of mortality in Europe and the United States after lung cancer [1][2]. Additionally, according to the American Cancer Society, about 12% of women in the USA are prone to develop BC during their lifetime [3][4][5][6].
Triple-negative breast cancer (TNBC) is a less common type of BC. About 10–20% of BCs are TNBC. TNBC consists of cancer cells, which either do not express estrogen and progesterone receptors or produce the protein named HER2 These cancers tend to be more common in women younger than the age of 40, who are usually African American [7].
Human genes BRCA1 and BRCA2 produce tumor suppressor proteins. These proteins participate in damaged DNA repairing and, therefore, play a crucial role in ensuring the stability of each cell’s genetic material. As a result, cells are more likely to develop additional genetic alterations, which may lead to cancer. BRCA2 genes, which are reported to increase the risk of female TNBC [8][9].
It is usually a combination of surgery, radiotherapy and chemotherapy. Therefore, hormone therapy is largely ineffective for treatment purposes. However, chemotherapy can cause various serious adverse effects such as cardiotoxicity, myelosuppression, alopecia and gastrointestinal problems [10]. Except for toxicity, drug resistance to chemotherapy is another major problem.
Major effort has been devoted by researchers in order to classify TNBC. Technology has facilitated researchers to analyze numerous data to compare different TNBCs and classify them in subgroups based on their similarities. Several ways of categorization of TNBCs have been reported such as the molecular classification, the immune classification, the classification based on differential prognosis, based on the cell type ambulating in the tumor environment, based on the presence or absence of androgen receptors or based on cellular type [11].
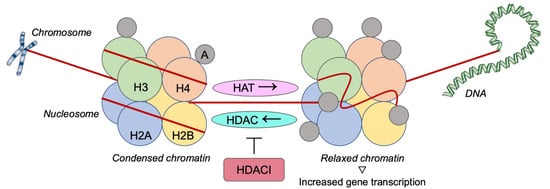
Given that the progression of cancer is often controlled via epigenetic processes, there is a growing interest in research focusing on mechanisms, genes and signaling pathways related to carcinogenesis with epigenetic modulation of gene expression. For example, histone deacetylases (HDACs) have a significant impact on chromatin remodeling and epigenetics. Therefore, their inhibitors consist of an appealing field for targeted therapy against BC and are widely studied [12][13]].
Along the same line with HDACs, numerous studies and both clinical and laboratory trials are taking place, in order to provide new targets and improve prognosis for TNBC. PARP inhibitors, the PI3K/AMT/TOR pathway, anti-angiogenetic factors, as well as immunotherapy are potential targets for the treatment of TNBC. This current review presents up-to-date studies, focusing on the progress made in the field of targeted therapies for TNBC.
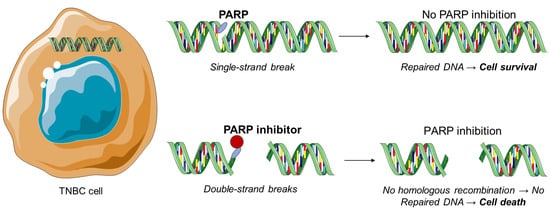
1.1. PARP Inhibitors
2. Investigational Drug Treatments for Triple-Negative Breast Cancer
2.1. Aurora Kinase Inhibitors
2.2. Histone Deacetylase Inhibitors
2.3. Other Inhibitors
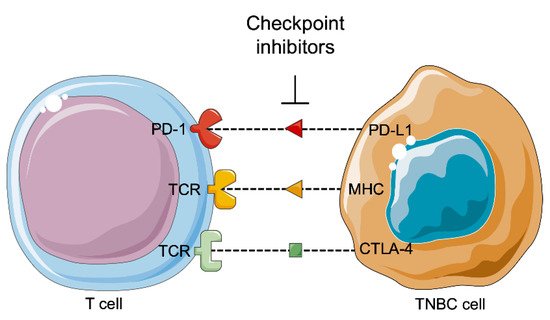
2.4. Immunotherapy
3. Conclusions
References
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac. J. Cancer Prev. APJCP 2016, 17, 43–46.
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988.
- Antoniou, A.C.; Easton, D.F. Models of genetic susceptibility to breast cancer. Oncogene 2006, 25, 5898–5905.
- Rivenbark, A.G.; O’Connor, S.M.; Coleman, W.B. Molecular and cellular heterogeneity in breast cancer: Challenges for personalized medicine. Am. J. Pathol. 2013, 183, 1113–1124.
- Nyante, S.J.; Lee, S.S.; Benefield, T.S.; Hoots, T.N.; Henderson, L.M. The association between mammographic calcifications and breast cancer prognostic factors in a population-based registry cohort. Cancer 2017, 123, 219–227.
- DeSantis, C.E.; Bray, F.; Ferlay, J.; Lortet-Tieulent, J.; Anderson, B.O.; Jemal, A. International variation in female breast cancer incidence and mortality rates. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1495–1506.
- Damaskos, C.; Garmpi, A.; Nikolettos, K.; Vavourakis, M.; Diamantis, E.; Patsouras, A.; Farmaki, P.; Nonni, A.; Dimitroulis, D.; Mantas, D.; et al. Triple-negative breast cancer: The progress of targeted therapies and future tendencies. Anticancer Res. 2019, 39, 5285–5296.
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017, 317, 2402–2416.
- Tai, Y.C.; Domchek, S.; Parmigiani, G.; Chen, S. Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. J. Natl. Cancer Inst. 2007, 99, 1811–1814.
- O’Brien, M.E.; Wigler, N.; Inbar, M.; Rosso, R.; Grischke, E.; Santoro, A.; Catane, R.; Kieback, D.G.; Tomczak, P.; Ackland, S.P.; et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol. 2004, 15, 440–449.
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Nikolettos, K.; Dimitroulis, D.; Diamantis, E.; Farmaki, P.; Patsouras, A.; Voutyritsa, E.; Syllaios, A.; et al. Molecular classification and future therapeutic challenges of triple-negative breast cancer. In Vivo 2020, 34, 1715–1727.
- Damaskos, C.; Garmpis, N.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; et al. Histone deacetylase inhibitors: An attractive therapeutic strategy against breast cancer. Anticancer Res. 2017, 37, 35–46.
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Kalampokas, E.; Kalampokas, T.; Spartalis, E.; Daskalopoulou, A.; Valsami, S.; Kontos, M.; Nonni, A.; et al. Histone deacetylases as new therapeutic targets in triple-negative breast cancer: Progress and promises. Cancer Genom. Proteom. 2017, 14, 299–313.
- Park, S.R.; Chen, A. Poly(Adenosine diphosphate-ribose) polymerase inhibitors in cancer treatment. Hematol. Oncol. Clin. N. Am. 2012, 26, 649–670.
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146.
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24.
- Eustermann, S.; Wu, W.F.; Langelier, M.F.; Yang, J.C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol. Cell 2015, 60, 742–754.
- Dawicki-McKenna, J.M.; Langelier, M.F.; DeNizio, J.E.; Riccio, A.A.; Cao, C.D.; Karch, K.R.; McCauley, M.; Steffen, J.D.; Black, B.E.; Pascal, J.M. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell 2015, 60, 755–768.
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Jovanović, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A randomized phase II neoadjuvant study of cisplatin, paclitaxel with or without everolimus in patients with stage II/III triple negative breast cancer (TNBC): Responses and long-term outcome correlated with increased frequency of DNA damage response gene mutations, TNBC subtype, AR status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045.
- Llombart-Cussac, A.; Bermejo, B.; Villanueva, C.; Delaloge, S.; Morales, S.; Balmaña, J.; Amillano, K.; Bonnefoi, H.; Casas, A.; Manso, L.; et al. SOLTI NeoPARP: A phase II randomized study of two schedules of iniparib plus paclitaxel versus paclitaxel alone as neoadjuvant therapy in patients with triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 154, 351–357.
- Patel, A.G.; De Lorenzo, S.B.; Flatten, K.S.; Poirier, G.G.; Kaufmann, S.H. Failure of iniparib to inhibit poly(ADP-Ribose) polymerase in vitro. Clin. Cancer Res. 2012, 18, 1655–1662.
- Kummar, S.; Wade, J.L.; Oza, A.M.; Sullivan, D.; Chen, A.P.; Gandara, D.R.; Ji, J.; Kinders, R.J.; Wang, L.; Allen, D.; et al. Randomized phase II trial of cyclophosphamide and the oral poly (ADP-ribose) polymerase inhibitor veliparib in patients with recurrent, advanced triple-negative breast cancer. Investig. New Drugs 2016, 34, 355–363.
- Evans, K.W.; Yuca, E.; Akcakanat, A.; Scott, S.M.; Arango, N.P.; Zheng, X.; Chen, K.; Tapia, C.; Tarco, E.; Eterovic, A.K.; et al. A population of heterogeneous breast cancer patient-derived xenografts demonstrate broad activity of PARP inhibitor in BRCA1/2 wild-type tumors. Clin. Cancer Res. 2017, 23, 6468–6477.
- Pothuri, B.; Brodsky, A.L.; Sparano, J.A.; Blank, S.V.; Kim, M.; Hershman, D.L.; Tiersten, A.; Kiesel, B.F.; Beumer, J.H.; Liebes, L.; et al. Phase I and pharmacokinetic study of veliparib, a PARP inhibitor, and pegylated liposomal doxorubicin (PLD) in recurrent gynecologic cancer and triple negative breast cancer with long-term follow-up. Cancer Chemother. Pharmacol. 2020, 85, 741–751.
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. 2021, 32, 240–249.
- Bischoff, J.R.; Anderson, L.; Zhu, Y.; Mossie, K.; Ng, L.; Souza, B.; Schryver, B.; Flanagan, P.; Clairvoyant, F.; Ginther, C.; et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998, 17, 3052–3065.
- Carmena, M.; Earnshaw, W.C. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854.
- Giet, R.; Prigent, C. Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine-threonine kinases. J. Cell Sci. 1999, 112, 3591–3601.
- Tayyar, Y.; Jubair, L.; Fallaha, S.; McMillan, N.A.J. Critical risk-benefit assessment of the novel anti-cancer aurora a kinase inhibitor alisertib (MLN8237): A comprehensive review of the clinical data. Crit. Rev. Oncol. Hematol. 2017, 119, 59–65.
- Girdler, F.; Gascoigne, K.E.; Eyers, P.A.; Hartmuth, S.; Crafter, C.; Foote, K.M.; Keen, N.J.; Taylor, S.S. Validating aurora B as an anti-cancer drug target. J. Cell Sci. 2006, 119, 3664–3675.
- Carpinelli, P.; Moll, J. Aurora kinase inhibitors: Identification and preclinical validation of their biomarkers. Expert Opin. Ther. Targets 2008, 12, 69–80.
- Gautschi, O.; Heighway, J.; Mack, P.C.; Purnell, P.R.; Lara, P.N., Jr.; Gandara, D.R. Aurora kinases as anticancer drug targets. Clin. Cancer Res. 2008, 14, 1639–1648.
- Vader, G.; Medema, R.H.; Lens, S.M. The chromosomal passenger complex: Guiding aurora-B through mitosis. J. Cell Biol. 2006, 173, 833–837.
- Ulisse, S.; Delcros, J.G.; Baldini, E.; Toller, M.; Curcio, F.; Giacomelli, L.; Prigent, C.; Ambesi-Impiombato, F.S.; D’Armiento, M.; Arlot-Bonnemains, Y. Expression of aurora kinases in human thyroid carcinoma cell lines and tissues. Int. J. Cancer 2006, 119, 275–282.
- Bernard, M.; Sanseau, P.; Henry, C.; Couturier, A.; Prigent, C. Cloning of STK13, a third human protein kinase related to Drosophila aurora and budding yeast Ipl1 that maps on chromosome 19q13.3-ter. Genomics 1998, 53, 406–409.
- Kimura, M.; Matsuda, Y.; Yoshioka, T.; Okano, Y. Cell cycle-dependent expression and centrosome localization of a third human aurora/Ipl1-related protein kinase, AIK3. J. Biol. Chem. 1999, 274, 7334–7340.
- Yang, K.T.; Li, S.K.; Chang, C.C.; Tang, C.J.; Lin, Y.N.; Lee, S.C.; Tang, T.K. Aurora-C kinase deficiency causes cytokinesis failure in meiosis-I and production of large polyploid oocytes in mouse. Mol. Biol. Cell 2010, 21, 2371–2383.
- Huck, J.J.; Zhang, M.; Jerome Mettetal, J.; Chakravarty, A.; Venkatakrishnan, K.; Zhou, X.; Kleinfield, R.; Hyer, M.L.; Kannan, K.; Shinde, V.; et al. Translational exposure-efficacy modeling to optimize the dose and schedule of taxanes combined with the investigational aurora A kinase inhibitor MLN8237 (Alisertib). Mol. Cancer Ther. 2014, 139, 2170–2183.
- Carducci, M.; Shaheen, M.; Markman, B.; Hurvitz, S.; Mahadevan, D.; Kotasek, D.; Goodman, O.B., Jr.; Rasmussen, E.; Chow, V.; Juan, G.; et al. A phase 1, first-in-human study of AMG 900, an orally administered pan-Aurora kinase inhibitor, in adult patients with advanced solid tumors. Investig. New Drugs 2018, 36, 1060–1071.
- Tolcher, A.W.; Kurzrock, R.; Valero, V.; Gonzalez, R.; Heist, R.S.; Tan, A.R.; Means-Powell, J.; Werner, T.L.; Becerra, C.; Wang, C.; et al. Phase I dose-escalation trial of the oral AKT inhibitor uprosertib in combination with the oral MEK1/MEK2 inhibitor trametinib in patients with solid tumors. Cancer Chemother. Pharmacol. 2020, 85, 673–683.
- Grant, S.; Dai, Y. Histone deacetylase inhibitors and rational combination therapies. Adv. Cancer Res. 2012, 116, 199–237.
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436.
- Cao, Z.A.; Bass, K.E.; Balasubramaniam, S.; Liu, L.; Schultz, B.; Verner, E.; Dai, Y.; Molina, R.A.; Davis, J.R.; Misialek, S.; et al. CRA-026440: A potent, broad spectrum, hydroxamic histone deacetylase inhibitor with antiproliferative and antiangiogenic activity in vitro and in vivo. Mol. Cancer Ther. 2006, 5, 1693–1701.
- Garmpi, A.; Garmpis, N.; Damaskos, C.; Valsami, S.; Spartalis, E.; Lavaris, A.; Patelis, N.; Margonis, G.A.; Apostolou, K.G.; Spartalis, M.; et al. Histone deacetylase inhibitors as a new anticancer option: How far can we go with expectations? J. BUON 2018, 23, 846–861.
- Leoni, F.; Fossati, G.; Lewis, E.C.; Lee, J.K.; Porro, G.; Pagani, P.; Modena, D.; Moras, M.L.; Pozzi, P.; Reznikov, L.L.; et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol. Med. 2005, 11, 1–15.
- Min, A.; Im, S.A.; Kim, D.K.; Song, S.H.; Kim, H.J.; Lee, K.H.; Kim, T.Y.; Han, S.W.; Oh, D.Y.; Kim, T.Y.; et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015, 17, 33.
- Ono, H.; Sowa, Y.; Horinaka, M.; Iizumi, Y.; Watanabe, M.; Morita, M.; Nishimoto, E.; Taguchi, T.; Sakai, T. The histone deacetylase inhibitor OBP-801 and eribulin synergistically inhibit the growth of triple-negative breast cancer cells with the suppression of survivin, Bcl-xL, and the MAPK pathway. Breast Cancer Res. Treat. 2018, 171, 43–52.
- Song, X.; Wu, J.Q.; Yu, X.F.; Yang, X.S.; Yang, Y. Trichostatin A inhibits proliferation of triple negative breast cancer cells by inducing cell cycle arrest and apoptosis. Neoplasma 2018, 65, 898–906.
- Maiti, A.; Qi, Q.; Peng, X.; Yan, L.; Takabe, K.; Hait, N.C. Class I histone deacetylase inhibitor suppresses vasculogenic mimicry by enhancing the expression of tumor suppressor and anti-angiogenesis genes in aggressive human TNBC cells. Int. J. Oncol. 2019, 55, 116–130.
- Milazzo, F.M.; Vesci, L.; Anastasi, A.M.; Chiapparino, C.; Rosi, A.; Giannini, G.; Taddei, M.; Cini, E.; Faltoni, V.; Petricci, E.; et al. ErbB2 targeted epigenetic modulation: Anti-tumor efficacy of the ADC trastuzumab-HDACi ST8176AA1. Front. Oncol. 2020, 9, 1534.
- Schmidt, H.B.; Görlich, D. Transport selectivity of nuclear pores, phase separation, and membraneless organelles. Trends Biochem. Sci. 2016, 41, 46–61.
- Görlich, D.; Mattaj, I.W. Nucleocytoplasmic transport. Science 1996, 271, 1513–1518.
- Jamali, T.; Jamali, Y.; Mehrbod, M.; Mofrad, M.R.K. Nuclear pore complex: Biochemistry and biophysics of nucleocytoplasmic transport in health and disease. Int. Rev. Cell Mol. Biol. 2011, 287, 233–286.
- Sun, Q.; Chen, X.; Zhou, Q.; Burstein, E.; Yang, S.; Jia, D. Inhibiting cancer cell hallmark features through nuclear export inhibition. Signal Transduct. Target.Ther. 2016, 1, 16010.
- Das, A.; Wei, G.; Parikh, K.; Liu, D. Selective inhibitors of nuclear export (SINE) in hematological malignancies. Exp. Hematol. Oncol. 2015, 4, 7.
- Arango, N.P.; Yuca, E.; Zhao, M.; Evans, K.W.; Scott, S.; Kim, C.; Gonzalez-Angulo, A.M.; Janku, F.; Ueno, N.T.; Tripathy, D.; et al. Selinexor (KPT-330) demonstrates antitumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res. 2017, 19, 93.
- Porter, P.L.; Malone, K.E.; Heagerty, P.J.; Alexander, G.M.; Firpo, E.J.; Daling, J.R.; Roberts, J.M. Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat. Med. 1997, 3, 222–225.
- Keyomarsi, K.; Tucker, S.L.; Buchholz, T.A.; Callister, M.; Ding, Y.; Hortobagyi, G.N.; Bedrosian, I.; Knickerbocker, C.; Toyofuku, W.; Lowe, M.; et al. Cyclin E and survival in patients with breast cancer. N. Engl. J. Med. 2002, 347, 1566–1575.
- Kallakury, B.V.; Sheehan, C.E.; Ambros, R.A.; Fisher, H.A.; Kaufman, R.P.; Ross, J.S. The prognostic significance of p34cdc2 and cyclin D1 protein expression in prostate adenocarcinoma. Cancer 1997, 80, 753–763.
- Soria, J.C.; Jang, S.J.; Khuri, F.R.; Hassan, K.; Liu, D.; Hong, W.K.; Mao, L. Overexpression of cyclin B1 in early-stage non-small lung cancer and its clinical implications. Cancer Res. 2000, 60, 4000–4004.
- Takerno, S.; Noguchi, T.; Kikuchi, R.; Uchida, Y.; Yokoyama, S.; Muller, W. Prognostic value of cyclin B1 in patients with esophageal squamous cell carcinoma. Cancer 2002, 94, 2874–2881.
- Mitri, Z.; Karakas, C.; Wei, C.; Briones, B.; Simmons, H.; Ibrahim, N.; Alvarez, R.; Murray, J.L.; Keyomarsi, K.; Moulder, S. A phase 1 study with dose expansion of the CDK inhibitor dinaciclib (SCH 727965) in combination with epirubicin in patients with metastatic triple negative breast cancer. Investig. New Drugs 2015, 33, 890–894.
- Krause, D.S.; Van Etten, R.A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005, 353, 172–187.
- Tolaney, S.M.; Tan, S.; Guo, H.; Barry, W.; Van Allen, E.; Wagle, N.; Brock, J.; Larrabee, K.; Paweletz, C.; Ivanova, E.; et al. Phase II study of tivantinib (ARQ 197) in patients with metastatic triple-negative breast cancer. Investig. New Drugs 2015, 33, 1108–1114.
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309.
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2006, 23, 1011–1027.
- Pham, E.; Yin, M.; Peters, C.G.; Lee, C.R.; Brown, D.; Xu, P.; Man, S.; Jayaraman, L.; Rohde, E.; Chow, A.; et al. Preclinical efficacy of bevacizumab with CRLX101, an investigational nanoparticle-drug conjugate, in treatment of metastatic triple-negative breast cancer. Cancer Res. 2016, 76, 4493–4503.
- Herbst, R.S.; Shin, D.M. Monoclonal antibodies to target epidermal growth factor receptor-positive tumors: A new paradigm for cancer therapy. Cancer 2002, 94, 1593–1611.
- Brabender, J.; Danenberg, K.D.; Metzger, R.; Schneider, P.M.; Park, J.; Salonga, D.; Hölscher, A.H.; Danenberg, P.V. Epidermal growth factor receptor and HER2-neu mRNA expression in non-small cell lung cancer is correlated with survival. Clin. Cancer Res. 2001, 7, 1850–1855.
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signaling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137.
- Mendelsohn, J. The epidermal growth factor as a target for cancer therapy. Endocr. Relat. Cancer 2001, 8, 3–9.
- Brinkman, A.M.; Chen, G.; Wang, Y.; Hedman, C.J.; Sherer, N.M.; Havighurst, T.C.; Gong, S.; Xu, W. Aminoflavone-loaded EGFR-targeted unimolecular micelle nanoparticles exhibit anti-cancer effects in triple negative breast cancer. Biomaterials 2016, 101, 20–31.
- Wali, V.B.; Langdon, C.G.; Held, M.A.; Platt, J.T.; Patwardhan, G.A.; Safonov, A.; Aktas, B.; Pusztai, L.; Stern, D.F.; Hatzis, C. Systematic drug screening identifies tractable targeted combination therapies in triple-negative breast cancer. Cancer Res. 2017, 77, 566–578.
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors-molecular basis and current perspectives in multiple myeloma. J. Cell Mol. Med. 2014, 18, 947–961.
- Rinnerthaler, G.; Gampenrieder, S.P.; Petzer, A.; Burgstaller, S.; Fuchs, D.; Rossmann, D.; Balic, M.; Egle, D.; Rumpold, H.; Singer, C.F.; et al. Ixazomib in combination with carboplatin in pretreated women with advanced triple-negative breast cancer, a phase I/II trial of the AGMT (AGMT MBC-10 Trial). BMC Cancer 2018, 18, 1074.
- Belkina, A.C.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477.
- Ali, I.; Choi, G.; Lee, K. BET inhibitors as anticancer agents: A patent review. Recent Pat. Anticancer Drug Discov. 2017, 12, 340–364.
- Liu, Z.; Wang, P.; Chen, H.; World, E.A.; Tian, B.; Brasier, A.R.; Zhou, J. Drug discovery targeting bromodomain-containing protein 4. J. Med. Chem. 2017, 60, 4533–4558.
- Park, I.H.; Yang, H.N.; Jeon, S.Y.; Hwang, J.A.; Kim, M.K.; Kong, S.Y.; Shim, S.H.; Lee, K.S. Anti-tumor activity of BET inhibitors in androgen-receptor-expressing triple-negative breast cancer. Sci. Rep. 2019, 9, 13305.
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107.
- Vetrivel, K.S.; Cheng, H.; Kim, S.H.; Chen, Y.; Barnes, N.Y.; Parent, A.T.; Sisodia, S.S.; Thinakaran, G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J. Biol. Chem. 2005, 280, 25892–25900.
- Golde, T.E.; Petrucelli, L.; Lewis, J. Targeting Abeta and tau in Alzheimer’s disease, an early interim report. Exp. Neurol. 2010, 223, 252–266.
- Sardesai, S.; Badawi, M.; Mrozek, E.; Morgan, E.; Phelps, M.; Stephens, J.; Wei, L.; Kassem, M.; Ling, Y.; Lustberg, M.; et al. A phase I study of an oral selective gamma secretase (GS) inhibitor RO4929097 in combination with neoadjuvant paclitaxel and carboplatin in triple negative breast cancer. Investig. New Drugs 2020, 38, 1400–1410.
- Brufsky, A.; Kim, S.B.; Zvirbule, Ž.; Eniu, A.; Mebis, J.; Sohn, J.H.; Wongchenko, M.; Chohan, S.; Amin, R.; Yan, Y.; et al. A phase II randomized trial of cobimetinib plus chemotherapy, with or without atezolizumab, as first-line treatment for patients with locally advanced or metastatic triple-negative breast cancer (COLET): Primary analysis. Ann. Oncol. 2021, 32, 652–660.
- Salimi, M. Future of triple negative breast cancer: Can immunotherapy treat this deadly subtype of breast cancer? Iran. Biomed. J. 2018, 22, 76–77.
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570.
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25.
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899.
- Nanda, R.; Chow, L.Q.M.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in patients with advanced triple-negative breast cancer: Phase Ib KEYNOTE-012 study. J. Clin. Oncol. 2016, 34, 2460–2467.
- Tolaney, S.M.; Ziehr, D.R.; Guo, H.; Ng, M.R.; Barry, W.T.; Higgins, M.J.; Isakoff, S.J.; Brock, J.E.; Ivanova, E.V.; Paweletz, C.P.; et al. Phase II and biomarker study of cabozantinib in metastatic triple-negative breast cancer patients. Oncologist 2017, 22, 25–32.
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018, 379, 2108–2121.
- Bernier, C.; Soliman, A.; Gravel, M.; Dankner, M.; Savage, P.; Petrecca, K.; Park, M.; Siegel, P.M.; Shore, G.C.; Roulston, A. DZ-2384 has a superior preclinical profile to taxanes for the treatment of triple-negative breast cancer and is synergistic with anti-CTLA-4 immunotherapy. Anticancer Drugs 2018, 29, 774–785.
- Santa-Maria, C.A.; Kato, T.; Park, J.H.; Kiyotani, K.; Rademaker, A.; Shah, A.N.; Gross, L.; Blanco, L.Z.; Jain, S.; Flaum, L.; et al. A pilot study of durvalumab and tremelimumab and immunogenomic dynamics in metastatic breast cancer. Oncotarget 2018, 9, 18985–18996.
- Cortés, J.; André, F.; Gonçalves, A.; Kümmel, S.; Martín, M.; Schmid, P.; Schuetz, F.; Swain, S.M.; Easton, V.; Pollex, E.; et al. IMpassion132 phase III trial: Atezolizumab and chemotherapy in early relapsing metastatic triple-negative breast cancer. Future Oncol. 2019, 15, 1951–1961.
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928.
- Winer, E.P.; Lipatov, O.; Im, S.A.; Goncalves, A.; Muñoz-Couselo, E.; Lee, K.S.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 499–511.


