Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bhaskar Saha | + 980 word(s) | 980 | 2021-06-21 11:02:21 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Saha, B.; Kubatzky, K. Ras Isoforms. Encyclopedia. Available online: https://encyclopedia.pub/entry/11442 (accessed on 07 February 2026).
Saha B, Kubatzky K. Ras Isoforms. Encyclopedia. Available at: https://encyclopedia.pub/entry/11442. Accessed February 07, 2026.
Saha, Bhaskar, Katharina Kubatzky. "Ras Isoforms" Encyclopedia, https://encyclopedia.pub/entry/11442 (accessed February 07, 2026).
Saha, B., & Kubatzky, K. (2021, June 29). Ras Isoforms. In Encyclopedia. https://encyclopedia.pub/entry/11442
Saha, Bhaskar and Katharina Kubatzky. "Ras Isoforms." Encyclopedia. Web. 29 June, 2021.
Copy Citation
The central protein in the oncogenic circuitry is the Ras GTPase that has been under intense scrutiny for the last four decades. The complexity of the Ras functioning is further exemplified by the fact that the three canonical Ras genes encode for four protein isoforms (H-Ras, K-Ras4A, K-Ras4B, and N-Ras).
Ras
signaling
therapy
Ras-isoforms
H-Ras
K-Ras
N-Ras
oncogene
cancer
1. Introduction
The cellular Ras GTPase serves as a crucial molecular switch for non-oncogenic and oncogenic circuitry in cells. It is a tightly regulated key signaling molecule for many cellular processes as diverse as proliferation, cell adhesion, migration, differentiation, and death (Figure 1). Novel structural and functional properties of Ras GTPase in controlling tubulogenesis in endothelial cells [1] and pseudopodium dynamics in Dictyostelium [2] are emerging. Since the discovery of the roles of Ras in oncogenesis in the 1980s, the development of small molecular inhibitors against Ras has been the prime focus of pharmaceutical companies.
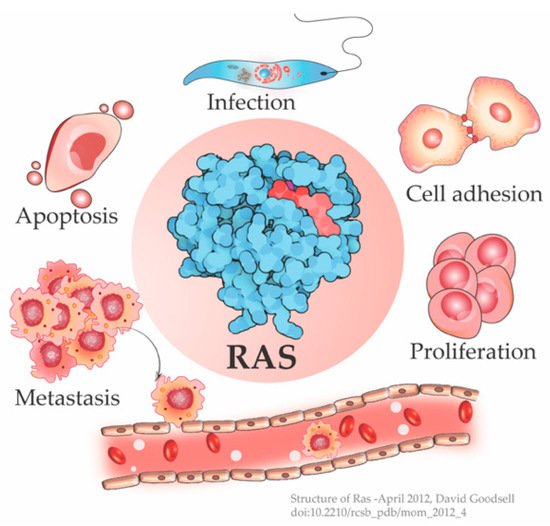
Figure 1. Ras is a signaling switch that regulates counteracting cellular functions like cell proliferation and apoptosis. Emerging studies have elucidated new functions of Ras e.g., in cytoskeletal rearrangement regulating cell adhesion and in parasitic infections like Leishmania, in addition to its well-known role in cancer progression. (Structure of Ras-April 2012, David Goodsell [3]).
2. The History of Ras Isoforms—From Viral Oncogenes to Pivotal Cellular Genes
In the 1960s, Jennifer Harvey observed that the viral preparation from leukemic rat induced sarcomas in newborn rats. This oncogenic viral genetic element inducing Rat Sarcoma was named H-Ras (Harvey Ras) [4]. Later, while serially passing Mouse Erythroblastosis virus (MEV) in Wister-Furth (W/Fu) rats, Kirsten identified another retrovirus carrying the Ras gene [5]. Initially named as a variant of Src (sarcoma), this gene was not only mutated in a diverse spectrum of cancers but also encoded a 21k Da protein that had crucial cellular roles in normal cells.
Scolnick et al. first hypothesized and Stehelin et al. later experimentally proved that these oncogenes were the proto-oncogenes which, when mutated and virally transferred, were converted to transforming oncogenes [6][7]. As observed by Harvey and Kirsten, the retroviruses arose as a result of the passage of the murine leukemia virus through rats. The process of genetic recombination accounts for the presence of such cellular genetic elements in simple retroviruses. A plausible hypothesis, as framed by various researchers, holds that integration of a provirus may occur upstream of a cellular sequence forming a chimera of cellular-viral genetic elements. In subsequent replications, non-homologous recombination occurs between the cellular and viral genetic elements leading to the acquisition and incorporation of cellular genetic elements into the retroviruses [8][9][10]. Later in the 1980s, Scolnick and his colleagues found the cellular origin of this membrane-associated protein, which is dependent on guanosine-5′-triphosphate (GTP) binding for its activation [11][12][13]. It was discovered that three loci of this gene encode for four proteins with ~80% sequence similarity: Harvey Ras (H-Ras), K-Ras A and B (Kirsten-Ras), and neuroblastoma Ras (N-Ras). K-Ras gene encodes for two proteins K-Ras4A and K-Ras4B via alternative splicing. These splice variants have distinct membrane targeting sequences. Except for a few residues, these isoforms have identical amino acid (aa) sequences in their G-domain (aa 1–86), and variations lie in the allosteric lobe (aa 87–166) and the hypervariable region (HVR) (aa 166–178/179) [14]. The HVR can be further subdivided into a linker domain (aa 166–178/179) and a targeting domain, wherein the post-translationally modified residues lie (aa 179/180–189) [15]. As the fourth exon in K-Ras encodes for the HVR, there lie differences in the HVR of K-Ras4A and K-Ras4B. K-Ras4A undergoes palmitoylation, whereas, K-Ras4B, which lacks a palmitoylation site, adds polylysine residues (Figure 2).
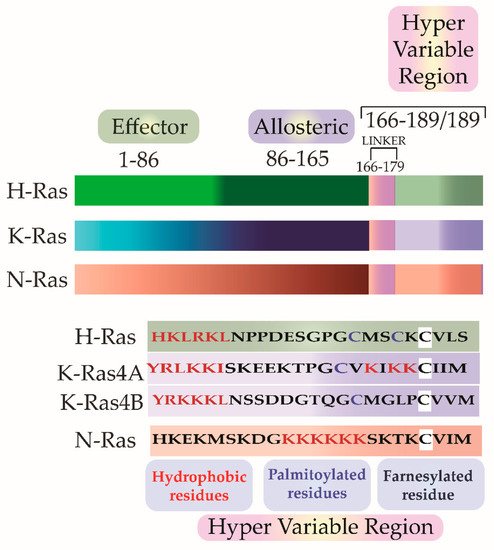
Figure 2. The isoforms share sequence similarities in their G-Domain (1–165 aa). This is followed by a Hyper Variable Region (HVR) (166–188/189), which differs in all three isoforms in sequence as well as in the post-translational modifications. In addition to farnesylation, a post-translational modification that all the three isoforms undergo, H-Ras and N-Ras further undergo dual and single palmitoylation, respectively. K-Ras undergo the addition of a stretch of polylysine residues.
3. Ras the Crucial Signal Relay Protein
The expression pattern of Ras isoforms is tissue-specific as well as developmental stage-specific [16][17]. The primary role of Ras emerged as a protein that assembles signaling complexes and relays signals to regulate an array of cellular activities. Emerging roles show its involvement in maintaining the integrity of actin cytoskeleton, in cell adhesion and migration, endocytosis, etc. [18]. This brings us to the crucial question—‘How does one signaling protein regulate counteracting processes like proliferation and apoptosis?’
Ras is a molecular switch that is activated by guanine nucleotide exchange factors (GEFs) that catalyze the exchange of guanosine diphosphate (GDP) with GTP. The counter process—the transition of Ras from active to the resting state—occurs by GTPase activating proteins (GAP) mediated GTP hydrolysis. Ras has a slow intrinsic GTP hydrolysis activity which is accelerated by GAPs by ∼105 folds [19]. Two well-characterized GAPs are neurofibromin (NF1) and Ras P21 Protein Activator 1 (RASA1) (p120GAP) [20]. If Ras deactivation fails, Ras remains at an active or “On” state. Once active, Ras binds to a range of effectors that carry out the downstream signaling. The specificity, as well as the diversity of signaling, arises due to binding with specific activators and effectors [21]. Chin et al. and Fisher et al. demonstrated that Ras-induced oncogenic transformation by uncontrolled cell proliferation requires a sustained expression of activated Ras and in an inducible oncogenic system, withdrawal of Ras expression led to tumor regression [22][23]. The ability of Ras to induce both proapoptotic and antiapoptotic signals may depend on an interplay between the type of receptor(s) activated, strength of activation, the access to activators and effectors, their binding specificities, kinetics, and stoichiometry of signaling. The outcome of Ras activation depends on whether it is a normal or an oncogenic cell, as a highly activated Ras in a normal cell will most likely relay a proapoptotic signal, whereas in an oncogenic setup it will relay an antiapoptotic signal [24].
References
- Norden, P.; Kim, D.J.; Barry, D.M.; Cleaver, O.B.; Davis, G.E. Cdc42 and k-Ras Control Endothelial Tubulogenesis through Apical Membrane and Cytoskeletal Polarization: Novel Stimulatory Roles for GTPase Effectors, the Small GTPases, Rac2 and Rap1b, and Inhibitory Influence of Arhgap31 and Rasa1. PLoS ONE 2016, 11, e0147758.
- van Haastert, P.J.; Keizer-Gunnink, I.; Kortholt, A. Coupled excitable Ras and F-actin activation mediates spontaneous pseudopod formation and directed cell movement. Mol. Biol. Cell. 2017, 28, 922–934.
- Structure of Ras-April 2012, David Goodsell. Available online: (accessed on 30 May 2021).
- Harvey, J.J. An unidentified virus which causes the rapid production of tumors in mice. Nature 1964, 204, 1104–1105.
- Kirsten, W.H.; Mayer, L.A. Morphologic Responses to a Murine Erythroblastosis Virus2. J. Natl. Cancer Inst. 1967, 39, 311–335.
- Scolnick, E.M.; Parks, W.P. Harvey sarcoma virus: A second murine type C sarcoma virus with rat genetic information. J. Virol. 1974, 13, 1211–1219.
- Stehelin, D.; Varmus, E.H.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nat. Cell Biol. 1976, 260, 170–173.
- Goldfarb, M.P.; Weinberg, R.A. Generation of novel, biologically active Harvey sarcoma viruses via apparent illegitimate recombination. J. Virol. 1981, 38, 136–150.
- Swanstrom, R.; Parker, R.C.; Varmus, H.E.; Bishop, J.M. Transduction of a cellular oncogene: The genesis of Rous sarcoma virus. Proc. Natl. Acad. Sci. USA 1983, 80, 2519–2523.
- Swain, A.; Coffin, J.M. Mechanism of transduction by retroviruses. Science 1992, 255, 841–845.
- Willingham, M.C.; Pastan, I.; Shih, T.Y.; Scolnick, E.M. Localization of the src gene product of the Harvey strain of MSV to plasma membrane of transformed cells by electron microscopic immunocytochemistry. Cell 1980, 19, 1005–1014.
- Ellis, R.W.; Defeo, D.; Shih, T.Y.; Gonda, M.A.; Young, H.A.; Tsuchida, N.; Lowy, D.R.; Scolnick, E.M. The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nat. Cell Biol. 1981, 292, 506–511.
- Gibbs, J.B.; Sigal, I.S.; Poe, M.; Scolnick, E.M. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc. Natl. Acad. Sci. USA 1984, 81, 5704–5708.
- Buhrman, G.; Holzapfel, G.; Fetics, S.; Mattos, C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc. Natl. Acad. Sci. USA 2010, 107, 4931–4936.
- Hancock, J.F.; Cadwallader, K.; Paterson, H.; Marshall, C.J. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991, 10, 4033–4039.
- Di, Y.; Schroeder, D.C.; Highfield, A.; Readman, J.W.; Jha, A.N. Tissue-specific expression of p53 and ras genes in response to the environmental genotoxicant benzo (α) pyrene in marine mussels. Environ. Sci. Technol. 2011, 45, 8974–8981.
- Leon, J.; Guerrero, I.; Pellicer, A. Differential expression of the ras gene family in mice. Mol. Cell. Biol. 1987, 7, 1535–1540.
- Pollock, C.B.; Shirasawa, S.; Sasazuki, T.; Kolch, W.; Dhillon, A.S. Oncogenic K-RAS Is Required to Maintain Changes in Cytoskeletal Organization, Adhesion, and Motility in Colon Cancer Cells. Cancer Res. 2005, 65, 1244–1250.
- Gideon, P.; John, J.; Frech, M.; Lautwein, A.; Clark, R.; Scheffler, E.J.; Wittinghofer, A. Mutational and kinetic analyses of the GTPase-activating protein (GAP)-p21 interaction: The C-terminal domain of GAP is not sufficient for full activity. Mol. Cell. Biol. 1992, 12, 2050–2056.
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857.
- Nair, A.; Chakraborty, S.; Banerji, L.A.; Srivastava, A.; Navare, C.; Saha, B. Ras isoforms: Signaling specificities in CD40 pathway. Cell Commun. Signal. 2020, 18, 1–12.
- Chin, L.; Tam, A.; Pomerantz, J.; Wong, M.; Holash, J.; Bardeesy, N.; Shen, Q.; O’Hagan, R.; Pantginis, J.; Zhou, H.; et al. Essential role for oncogenic Ras in tumor maintenance. Nature 1999, 400, 468–472.
- Fisher, G.H.; Wellen, S.L.; Klimstra, D.; Lenczowski, J.M.; Tichelaar, J.W.; Lizak, M.J.; Whitsett, J.A.; Koretsky, A.; Varmus, H.E. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001, 15, 3249–3262.
- Cox, A.D.; Der, C.J. The dark side of Ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revision:
1 time
(View History)
Update Date:
29 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No