Intellectual disability (ID) is a condition characterized by limited intellectual functioning and adaptive behaviors. It affects 1–3% of the worldwide population and no pharmacological therapies are currently available. Too many genes have been found mutated in ID patients suggesting that the genetic bases are highly heterogeneous and apparently unrelated. Bibliomic analysis reveals that ID genes converge onto a few biological modules, one of these being cytoskeleton dynamics and Rho GTPases transduction. Genetic variants exert their effects at different levels in a hierarchical arrangement, starting from the molecular level and moving toward higher levels of organization, i.e., cell compartment and functions, circuits, cognition, and behavior. Thus, cytoskeleton alterations which have an impact on cell processes such as neuronal migration, neuritogenesis, and synaptic plasticity, rebound on the overall establishment of an effective network and consequently on the cognitive phenotype. Systems biology approaches are more focused on the overall interconnected network rather than on individual genes, encouraging the design of therapies that aim to correct common dysregulated biological processes. This review summarizes current knowledge about cytoskeleton control in neurons and its relevance for the ID pathogenesis, exploiting in silico modeling and translating the implications of those findings to biomedical research.
1. Introduction
ID is a heterogeneous group of neurodevelopmental disorders (NDDs), usually diagnosed before the age of 18, characterized by significant limitations in both intellectual functioning (IQ < 70) and adaptive behavior as expressed in conceptual, social, and practical adaptive skills
[1]. It affects 1–3% of the worldwide population, depending on the inclusion criteria, with a higher prevalence among males
[2][3][4][5]. Because of its high frequency, limited treatability, and required lifelong care, ID has a dramatic social and economic impact.
ID is manifested as both syndromic and non-syndromic forms, depending on whether the disability is associated with other symptoms, and it is clinically classified referring to severity (mild, moderate, and severe) and penetrance (partially to fully penetrant)
[1]. A phenotype-based cluster analysis was made by Kochinke et al., establishing gene–phenotype relationships and revealing compromised molecular processes that underlie specific ID subgroups. For example, genes involved in growth factor signaling pathways, such as the MAPK pathway, are enriched in comorbidities such as short stature and ectodermal anomalies as compared to all other ID-associated genes. Epilepsy, metabolic dysfunctions, and myopathy are instead co-occurring within a cluster of genes enriched with mitochondrial function. Microcephaly and behavioral abnormalities were linked to two clusters comprising genes enriched with chromatin-related functions.
ID mutations can be autosomal–recessive, autosomal–dominant (mostly de novo), or X-linked. The latter two are responsible for the higher prevalence of ID in males versus females.
The causes of ID are heterogeneous and still to be completely defined: it has been estimated that half of all cases are due to environmental factors such as intrauterine/neonatal insults (preterm-birth complications, intrapartum-related factors such as hypoxic-ischemic encephalopathy, and infections like meningitis and neonatal tetanus) and postnatal risk factors such as severe malnutrition during infancy. The other half of the cases are associated with genetic variants, highly heterogeneous, and only partially identified. According to the SysID database
[1], 1454 ID genes (excluding 1224 annotated as low-confidence ID genes) have been identified, some of which code for proteins involved in the Rho GTPases signaling pathway, such as
OPHN1 (oligophrenin 1),
ARHGEF9 (Cdc42 guanine nucleotide exchange factor 9),
FGD1 (FYVE, RhoGEF, and PH domain-containing 1),
RAC1 (Rac family small GTPase 1), and
PAK3 (P21-activated kinase 3).
Based on a wealth of experimental data from animal models and cultured neurons, it is widely accepted that cognitive deficits in ID patients are linked to altered neuronal networking, impaired synaptic plasticity, and excitation/inhibition unbalance in the cerebral cortex and hippocampus, resulting in abnormal information processing
[6][7][8][9][10][11].
2. Cytoskeleton Functions in Neuronal Development
During development, neurons migrate to find synaptic partners and establish the complexity of the neuronal wiring. Neurite extension and navigation are possible thanks to the formation of the growth cone, a sensory-motile structure at the tip of the growing axon directed by chemotaxis
[12]. The structure of the growth cone is characterized by a dynamic periphery, in which actin filaments extend and retract to explore the surrounding environment, and by a more stable center that forms the axonal shaft
[13]. The interplay between microtubule assembly and actin dynamics is then essential for axonal elongation. The polarity of microtubules, essential for the directional transport of proteins and organelles
[14], allows the sliding movement that supports axon formation, as tubulin monomers are continuously transported at the leading edge of the growth cone
[15].
Microtubules’ and actin filaments’ polymerization result from the addition of α/β-tubulin and glomerular actin (G-actin), respectively
[16][17]. The rate of filament elongation and morphogenesis depends on the concentration and availability of monomers but also on the presence of proteins that regulate the assembly/disassembly kinetics and those responsible for increasing the level of complexity for higher-order network structures
[18][19]. While microtubules are the stiffest components of the cytoskeleton and can switch between a stably growing state and a rapidly shrinking one
[20], actin filaments are less rigid and more organized, supporting the overall structure and allowing the motility of the leading edge
[21].
It is assumed that growth cones are already provided with all the proteins necessary for synaptogenesis during their searching for contacts
[22]. The protrusion of filopodia finger-like structures retract upon contact with the postsynaptic cell to form a vestigial presynaptic terminal. These filopodia are characterized by a less tight bundle that is more dynamic compared to the architectural stability of conventional filopodia
[23]. This dynamism is required to let the nascent spine be perfectly aligned between the presynaptic active zone and the postsynaptic density (PSD). The juxtaposition is possible thanks to the presence of cell adhesion molecules that provide perfect docking geometries between the two membranes in the synaptic cleft
[24]. The actin cytoskeleton is involved in spine morphogenesis, as it controls changes in spine shapes. Immature dendritic spines show linear and thin-like structures, but, after making contact with the presynaptic terminal, actin filaments begin to cluster and enlarge the contact surface to form a mature spine with a mushroom-like shape
[25]. This is conceivable thanks to actin-related proteins that generate branched filaments, such as the ARP2/3 complex, balanced with capping proteins, such as CAPZ, to restrict their elongation and actin severing proteins, such as ADF/cofilin, that enhance filament disassembly
[26]. In addition, several scaffold proteins contribute to the maturation of dendritic spines controlling actin dynamics, e.g., PSD95, SHANK, and SRCIN1 (also known as p140Cap)
[27][28][29].
Synapses are not static formations. They undergo changes in postnatal life, while carrying out specific activities, e.g., learning, and in specific periods, e.g., synaptic pruning during adolescence. In particular, synaptic plasticity takes place in an activity-dependent manner: LTP is the result of strengthened synapses after high-frequency stimulation from the presynaptic terminal, while long-term depression (LTD) is a decrease in synaptic activity after low-frequency signals
[30]. For LTP, the presence of NMDA-type glutamate receptors in the membrane of the postsynaptic cells allows the insertion of new AMPA receptors in response to high-frequency stimuli. The localization of these ionotropic, excitatory glutamate receptors leads to an increase in the postsynaptic current and consequently to a stronger connection in a positive feedback loop. Both AMPA receptors’ and NMDA receptors’ trafficking relies on the actin cytoskeleton
[31][32][33][34].
2.1. The Core Regulation of Actin Dynamics
Alterations in neurites and spine morphology, as well as in neuronal migration properties, have been consistently associated with ID and other NDDs that include ID as a main and recurrent phenotype
[35]. These developmental features rely on the proper actin cytoskeleton dynamics, as neurite outgrowth, axonal migration, synaptogenesis, and synaptic plasticity are the result of three main processes: fibrous-actin (F-actin) dynamics (elongation/severing/branching), actin–myosin contractility, and F-actin coupling with the extracellular matrix
[36][37]. All three processes are regulated by a complex protein network in which the Rho-family small GTPases RAC1, CDC42, and RHOA emerge as hubs (A).
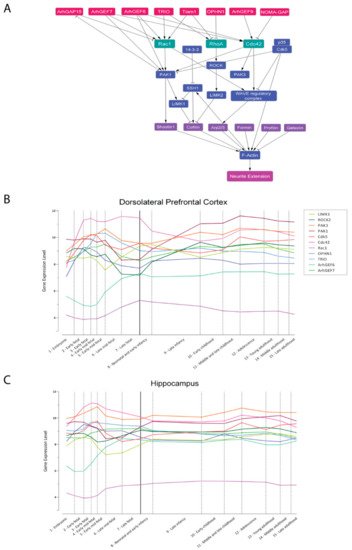
Figure 2. Proteins involved in the regulation of neurite elongation. (
A). PPI network of the best-characterized components of the Rho GTPase signaling RAC1, RHOA, and CDC42 realized with Cytoscape
[38]. Boxes represent the nodes (proteins), while the arrows indicate the edges (interactions). GTPases are reported in green, their GEFs and GAPs in red, their effectors in blue, and actin-binding or actin-modifying proteins in purple. Edges can be either “activatory” (arrowheads) or “inhibitory” (blunted lines). The “neurite elongation” node represents the phenotypic outcome. Acronyms are spelled out in the text. (
B,
C). Expression trajectories of ID-related genes in the human dorsolateral prefrontal cortex (
B) and hippocampus (
C). Quantile normalized gene-level expression values (log
2 transformed) inferred from Human Brain Transcriptome database
[39] were plotted against logarithmic age in days. The pattern was summarized by the smoothed curves of the expression values. Dashed lines divide periods of development and the solid line separates prenatal from postnatal periods. Individual genes are color-coded, legend in panel (
C).
This section illustrates in detail the key components of the signaling pathway responsible for the control of the dynamics of the actin cytoskeleton, focusing on the biochemical and cellular role of each protein and its links with neurological and cognitive deficits in human and animal models.
2.2. Synaptogenesis and Synaptic Plasticity
A set of mutations identified in ID specifically affect spine development and morphological changes during maturation. In this section, we summarize the most relevant findings.
KALRN: KALRN (kalirin) is a GEF for RAC1
[40]. In mice, KALRN expression increases at two weeks of age, a key time for synaptogenesis
[41].
Kalrn-KO mice showed decreased synaptic density in the apical dendrites of CA1 hippocampal neurons, along with learning deficits
[42].
PPP1R9A and PPP1R9B: PPP1R9A (neurabin -I) and PPP1R9B (also called neurabin-II or spinophilin) show F-actin cross-linking and phosphatase activity. They are enriched in dendritic spines
[43] and are likely to influence dendritic spine morphology and function through their interaction with F-actin
[44].
Ppp1r9b-KO mice have a reduced number of filopodia and an increase in spine density
[45].
ARHGEF2: Neurabin-I and Neurabin-II interact with the Rho GEF ARHGEF2 (also known as Lfc). This interaction selectively regulates Rho-dependent organization of F-actin in spines; ARHGEF2 is maintained inactive/sequestered through the interaction with microtubules and targeted to dendritic spines as a result of the interaction with Neurabin-I and Neurabin-II
[46].
ARHGEF2 mutation leads to reduced activity of the RHOA pathway. A homozygous frameshift mutation in the
ARHGEF2 is associated with ID, midbrain–hindbrain malformation, and mild microcephaly in a consanguineous family of Kurdish–Turkish descent
[47].
SHANK: SHANK family proteins are higher-order organizing scaffold proteins of the PSD. They are known to interact with the ABP DBNL (drebrin like) in the PSD to promote the reorganization of actin after stimulation
[48]. SHANK proteins activate RAC1 signaling at the PSD by recruiting ARHGEF7 through its PDZ C-terminal domain
[49]. Indeed, heterozygous mice lacking a SHANK3 C-terminus have an impaired actin polymerization and a consequent decrease of NMDA receptor delivery to the postsynaptic plasma membrane
[50]. SHANK3 can also directly interact with the ARP2/3 complex to increase F-actin level by decapping the barbed ends of actin filaments, thus promoting filament extension
[51]. A large variety of alterations in SHANK proteins are grouped as “shankopathies” and are linked with NDDs characterized by alterations in the actin cytoskeleton network
[52].
It has been proposed that in the 22q13 deletion syndrome, an NDD characterized by ID, the disruption of
SHANK3 is responsible for the clinical disorder
[53][54][55].
CAMK2: The activity of CAMK2, which is stimulated upon the increase in intracellular calcium concentration, is essential for AMPA receptor delivery to the membrane of silent synapses and SYNGAP activation
[56][57]. Whole-exome sequencing identified 19 rare de novo variants of CAMK2A and CAMK2B in 24 unrelated ID patients
[58].
2.3. The Role of Microtubules in ID
Microtubules are basic elements of the cytoskeleton and actively participate in most neurodevelopmental processes. Neurons depend on microtubule dynamics for cell division, axon guidance, intracellular trafficking, and synapse formation
[59]. They are constituted by heterodimers of α- and β-tubulin that associate to form a hollow cylinder. The stable to dynamic microtubules ratio is significantly higher in the neurite that is meant to form the axon as compared to the other neurites, indicating that stable microtubules are essential for axon specification
[60][61]. Microtubules’ polarity is required to deliver various cargoes to the correct location within the cell to assure axonal trafficking and to maintain the correct neuronal morphology. Microtubules also play a crucial role in spinogenesis, as dynamic microtubules penetrate dendritic spines to modulate their morphology by interacting with a large variety of microtubule-associated proteins
[62][63][64]. Some of them act directly on microtubules to affect their assembly or stability, while others act indirectly by modulating tubulin level or intracellular transport; for example, severing proteins are essential to increase tubulin monomers’ availability and to reorganize microtubules’ scaffold architecture
[65], while microtubule plus-end tracking proteins (+TIPs) are responsible for the regulation of microtubules by interacting with the plus ends and by functioning as a scaffold for other regulatory proteins
[66][67]. Cytoplasmic linker proteins (CLIPs), a subgroup of +TIPs, are fundamental for microtubule invasion into the growth cone leading edge and into nascent dendritic spines
[68][69].
Many microtubule-associated genes are linked to ID and to other NDDs in which ID appears as a prominent and recurrent phenotype:
ADNP mutations are associated with ASD;
ASPM, MCPH1, STIL, CDK5RAP2, CENPJ, PRUNE1, and
KIF20 mutations are associated with microcephaly;
TUBB2B mutations are associated with polymicrogyria;
LIS1,
DCX, and
TUBA1A are linked to lissencephaly
[59].
KIF1A, KIF4A, KIF5C, and KIF7: Kinesins are motor proteins that move along microtubules in an anterograde fashion, transporting their cargo towards microtubules’ plus end. KIF1A (kinesin family member 1A) is selectively expressed in neurons, and its partial or total depletion results in the disruption of axonal and dendritic transports
[70]. A dominant de novo missense mutation in
KIF1A was found in a patient with non-syndromic ID
[71], and other de novo mutations were found in six patients affected by severe early-infantile neurodegenerative syndrome
[72]. Next-generation sequencing revealed mutations in other kinesin family members such as
KIF4A and
KIF5C for which the causative role in ID is supported by evidence obtained using KO models
[73]. Mutations in
KIF2A and
KIF5C were reported in patients with malformations of cortical development presenting ID
[74], and homozygous mutations in
KIF7 were found in patients with ciliary disorders in which ID is part of the phenotype
[75].
KIFBP: KIFBP (kinesin family binding protein) is a modulator of kinesins activity. Homozygous nonsense mutations of KIFBP are associated with Goldberg–Shprintzen syndrome, which is a form of syndromic ID
[76].
CHAMP1: CHAMP1 (chromosome alignment maintaining phosphoprotein 1) is a zinc finger protein that regulates chromosome segregation during mitosis. De novo
CHAMP1 mutations are associated with GDD and ID
[77][78][79].
CLIP1: CLIP1 (CAP-Gly Domain Containing Linker Protein 1) is a +TIP that regulates microtubule growth and bundling. Next-generation sequencing revealed an autosomal recessive form of ID associated with a nonsense variant in
CLIP1 in an Iranian consanguineous family
[80].
KATNAL1: KATNAL1 (katanin catalytic subunit A1 like 1) is one of the two major catalytic subunits of the microtubule-severing enzyme Katanin, together with KATNAL2. Three unrelated patients with heterozygous de novo deletions encompassing 13q12.3 presented moderate ID phenotype; since this region contains
KATNAL1, this gene has been proposed as a candidate ID gene
[81].
MID1 and MID2: MID1 (midline 1) and MID2 are E3 ubiquitin ligases that have a role in microtubule stability and organization.
MID1 mutations are associated with Opitz G/BBB syndrome, in which mild to severe ID can appear
[82]. A
MID2 missense mutation that disrupts its interaction with microtubules is associated with XLID
[83].
CDKL5: CDKL5 (cyclin-dependent kinase-like 5) is a kinase protein essential for brain development. CDKL5 interacts with IQGAP1 (IQ motif containing GTPase activating protein 1), through which it forms a functional complex with its effectors RAC1 and CLIP1, MAPRE2 (microtubule-associated protein RP/EB family member 2), MAP1S (microtubule-associated protein 1S), ARHGEF2, and SHTN1
[84][85][86], thus stressing its regulation over cytoskeleton dynamics, in particular over microtubules.
CDKL5 mutations cause the so-called CDKL5 deficiency disorder in which severe ID is one of the most important clinical manifestations
[87].
3. Concluding Remarks
Current treatments of ID are largely based on psychosocial measures, environmental enrichment, dedicated educational plans, and motor activity, while pharmacotherapies are lagging. Recent evidence suggests that some phenotypes associated with cognitive disabilities can be reversed, through either genetic approaches
[88] or pharmacotherapy. At present, whether these provide a realistic opportunity for treatment remains to be defined.
One obstacle is represented by the high genetic variability observed in ID. SB approaches and integrative tools are beginning to deconvolute and model the core biological processes responsible for the altered circuitry and synaptic dysfunctions in ID. One of these processes is the regulation of cytoskeleton dynamics, whose hubs are the small GTPases of the Rho class. A relevant outcome of our current knowledge is the possibility to leverage the wealth of experimental and mutational data to derive a computational model, through which we will learn more about the behavior of the system.
Rho class GTPases are promising targets for pharmacological intervention and can be positively or negatively modulated by acting upstream, on the regulatory partners, or downstream, on actin stability. To achieve a brain-specific, GTPase-specific, modest, and controlled remodulation, a full characterization of the PPI between GTPases and their regulatory partners is required. Current knowledge led to the identification of several potentially valid compounds, some of which are well characterized for their biological activity, while others still need a proper characterization.
Future efforts should focus on completing our knowledge about the cytoskeleton core regulatory network, considering that relevant elements might still be missing or have been overlooked. Proteomic analyses focused on the human neuronal cytoskeleton are needed, intending to identify novel druggable elements participating in the GTPase regulatory network or neurodevelopmental processes, i.e., neurite elongation, neuronal migration, and adhesion.
Since most of the current knowledge is based on animal models, an area of strong interest is the generation and validation of cellular models of ID based on human neurons. Such models should recapitulate the cortical and hippocampal endophenotype of human ID and offer the possibility to examine the excitatory/inhibitory balance. In light of these observations, human iPSCs offer a valid model to identify new valuable readouts and to start screening compounds able to alleviate ID
[89].
 +1 point
+1 point





