 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giorgio Tasca | + 2346 word(s) | 2346 | 2021-06-09 05:24:11 | | | |
| 2 | Lily Guo | Meta information modification | 2346 | 2021-06-13 04:57:50 | | | | |
| 3 | Lily Guo | Meta information modification | 2346 | 2021-06-13 04:58:20 | | | | |
| 4 | Nicola MOSCA | + 673 word(s) | 3019 | 2021-06-15 11:54:01 | | |
Video Upload Options
Reactive oxygen species are (ROS) are signaling molecules moderately and continuously produced by skeletal muscles as a consequence of their contractile activity and high mitochondrial oxygen consumption. The main source of ROS production is located in the cytosol through the activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX enzymes), xanthine oxidase (XO), and nitric oxide synthase (NOS), and by the mitochondrial electron transport chain. When ROS exceed the antioxidant buffering capacity of tissues, oxidative stress occurs.
1. Introduction
The oxidative damage caused by an overproduction of reactive oxygen species (ROS) to biomolecules may affect skeletal muscle homeostasis and functionality, thus exacerbating pathological conditions in hereditary myopathies and muscular dystrophies [1]. Indeed, muscle degeneration, mitochondrial dysfunction, inflammation, and insufficient muscle regeneration are strictly linked to oxidative stress, which stems from the imbalance between ROS generation and protection provided by antioxidants. When such a disequilibrium of redox homeostasis occurs, the first intracellular defense line is represented by the activation of the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), the master regulator of antioxidant response [2]. Many Nrf2 inducers have been reported to counteract oxidative stress in several physiological and pathological conditions, by upregulating the antioxidant defenses, inhibiting inflammation, and improving mitochondrial function [3][4].
Muscular dystrophies are a phenotypically and genotypically heterogeneous group of inherited muscular disorders with common histopathological features that include inflammation, degeneration, necrosis, and fibrosis. In addition to the pathogenic consequence arising from individual gene mutations, oxidative stress has been identified as one of the most relevant causes of muscle damage in these disorders [1][5][6].
This review summarizes the current knowledge about the role of oxidative stress in the pathogenesis of muscular dystrophies and highlights potential new targets for therapies.
2. The Role of Oxidative Stress in Muscle Inflammation
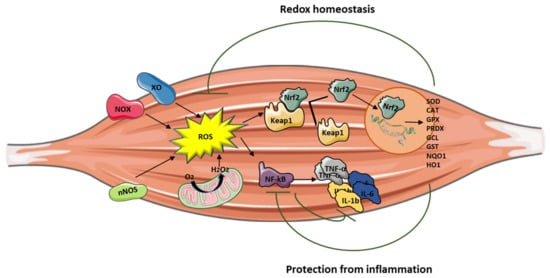
Inflammation can be triggered by an imbalance of cellular oxidative–antioxidant homeostasis. Usually, a moderate ROS production does not stimulate inflammation since ROS are mostly removed by cellular antioxidant systems. However, when ROS production, catalyzed by enzymes including NOX, COX-2, and XO, exceeds the threshold of redox balance, NF-κB is activated, thus triggering inflammation [7][8].
NF-κB is, therefore, the primary redox-sensitive signaling pathway of inflammation. The activation of protein kinase C (PKC) and NF-κB-induced kinase, a member of the MAPK family, is known to activate IKK, which phosphorylates the IκB subunit of the NF-κB complex and unleashes p50/p65 for nuclear binding [7][9]. As a result of NF-κB activation, proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6, are produced by immune cells and/or damaged muscle tissues, thus further promoting the expression of adhesion molecules, including vascular cell adhesion molecule Moreover, the NO production contributes to increase blood flow and the chemotactic effects of adhesion molecules, facilitating the infiltration of inflammatory cells into the affected area.
Mitochondria are also involved in the inflammatory process, and high levels of proinflammatory cytokines affect mitochondrial ROS generation and metabolic function. PGC-1α, which is a key regulator of muscle metabolism and mitochondrial biogenesis, also plays a role as an anti-inflammatory agent. Studies performed on PGC-1α KO mice models demonstrated high basal mRNA expression of TNF-α and IL-6 in skeletal muscle and in serum, showing that PGC-1α is able to effectively modulate local and systemic inflammation [10]. In addition, mice overexpressing PGC-1α showed low expression of TNF-α and IL-6 [11], suggesting that PGC-1α may play a protective role in the inflammatory response by reducing the generation of proinflammatory cytokines.
Nrf2 activation can attenuate inflammation by modulating the expression of NF-κB [12][13] (Figure 1). Nrf2 exerts its anti-inflammatory function via the activation of HO-1 that catalyzes the degradation of heme into carbon monoxide (CO), free iron (Fe2+), and biliverdin, before being converted to the antioxidant bilirubin [14][15][16]. Muscle damage and inflammation worsened after ablation of HO-1 in the mdx mouse [17], while pharmacological induction of HO-1 exerted a protective effect on skeletal muscle through the inhibition of NF-κB [18].
It is important to note that Nrf2 binds DNA in proximity of the genes encoding IL-1β and IL-18 to modulate their transcription [19], thereby supporting its direct inhibitory effect on NOD-like receptor protein 3 (NLRP3) inflammasome priming.
3. Oxidative Stress in Early Onset Myopathies
Early-onset myopathies are inherited muscle diseases occurring during infancy or early childhood. These can be further classified into congenital muscular dystrophies (CMDs) and congenital myopathies (CMs) [ 31,32]. They are typically associated with muscle weakness and hypotonia, delayed motor development, and difficult or absent ambulation, sometimes with orthopedic complications, respiratory and cardiac failure, and, in the most severe forms, premature death [20].
CMDs and CMs have been classified on the basis of their major muscle morphological features. Patients with CMDs present dystrophic lesions on muscle biopsies, with or without necrotic fibers and regeneration, whereas, in congenital myopathies, muscles are generally not dystrophic but show characteristic structural changes in the internal fiber architecture [21]. CMDs and CMs share several pathophysiological pathways, involving proteins essential for the embryonic muscle development, the complex architectural structure of muscle fibers, the excitation/contraction coupling, and the redox regulation system [21].
The deregulation of redox homeostasis is a mechanism implicated in the pathogenesis of early-onset myopathies, and significant progress has been reached in the understanding of these diseases through the analysis of oxidative-mediated processes [22][23][24][25]. Proteins involved in redox regulation represent hallmarks and potential therapeutic targets in several early-onset myopathies, including RYR1- and SEPN1-related myopathies, and in Duchenne muscular dystrophy (DMD) [26][27][28][29].
RYR1-related myopathies are a group of skeletal muscle disorders caused by mutations in the ryanodine receptor gene, RYR1 [30]. They are clinically characterized by a wide spectrum of symptoms ranging from slowly progressive hip and axial muscle proximal weakness, to malignant hyperthermia [31][32]. RYR1, also known as skeletal muscle calcium release channel, is an essential component of the excitation–contraction coupling apparatus and contains many redox-sensitive reactive cysteines, strongly involved in calcium handling [33][34][35][36][37]. The use of N-acetylcysteine (NAC), the precursor of the antioxidant glutathione (GSH), improved muscle function and restored myotube phenotype in RYR1 mutated cell cultures and in the Y522S mouse model, opening the way to clinical trials in patients [38][39][40][41].
Defects in redox homeostasis have also been described in SEPN1-related myopathy, an inherited muscle disease caused by mutations in the SELENON (or SEPN1) gene. Selenoproteins are a family of proteins containing several selenocysteine residues involved in modulating redox calcium homeostasis and protecting against oxidative stress [27]. SEPN1-mutated patients often show a severe weakness of neck and trunk muscle, leading to scoliosis, spinal rigidity, and life-threatening respiratory insufficiency [42]. Limited motility and body rigidity after forced swimming test were reported in Sepn1-deficient mouse models [43], and increased protein oxidation, abrogated after pretreating cells with NAC, was described in myoblasts primary cultures from patients [44], leading to NAC clinical trials (SELNAC, clinicaltrials.gov, identifier: NCT02505087).
Increasing evidence shows a central role for oxidative stress in DMD and Becker muscular dystrophy (BMD), the most common muscular dystrophy in childhood. DMD is a severe X-linked recessive neuromuscular disorder affecting one in 3600 boys [45]. Muscle degeneration results in weakness, delayed motor milestones, and, eventually, loss of ambulation. DMD is caused by mutations in the gene encoding for dystrophin, a membrane protein whose absence is responsible for increased susceptibility to damage, with myofiber necrosis and secondary inflammation as a result of skeletal muscle contraction [45][46][47].
Oxidative stress makes the dystrophin-deficient heart and skeletal muscle highly susceptible to injury, exacerbating the pathological features of the disease [48][49][50][51][52]. NOX is the main source of ROS in dystrophin-deficient models, contributing to enhanced Ca2+ influx and activation of the Src kinase [53]. Furthermore, decreased levels of the antioxidant GSH, with a concomitant increase of its oxidized form GSSG and a reduced activity of the protective enzyme GPX, have been found in dystrophic muscles and in peripheral blood of patients with DMD [54][55]. Notably, preclinical studies using antioxidant drugs have evidenced beneficial effects on dystrophin-deficient mouse models, such as reduced muscle damage, decreased necrosis, inflammation, and fibrosis [56][57][58][59][60][61].
4. Oxidative Stress in Adult-Onset Muscular Dystrophies
Adult-onset muscular dystrophies are inherited disorders with different severity, age of onset, and type of muscle involvement. Among them, the most prevalent is myotonic dystrophy type 1 (Steinert disease), Facioscapulohumeral muscular dystrophy, and limb-girdle muscular dystrophies.
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common forms of muscular dystrophy. It is characterized by a distinctive pattern of skeletal muscle weakness and a wide spectrum of disease severity. This contraction, together with a polyadenylation signal distal to the repeats, allows the stable transcription of the DUX4 retrogene that is normally repressed in somatic tissues including skeletal muscle. Moreover, DUX4 expression in skeletal muscle can also lead to activation of different genes involved in atrophy, protein degradation, and innate immunity, and it negatively regulates myogenesis [62][63].
Bosnakovski and colleagues showed that DUX4 alters the expression of genes implicated in redox balance and enhances the sensitivity of C2C12 myoblasts to pro-oxidant compounds, while antioxidant treatment reduced DUX4 toxicity [64][65]. Interestingly, oxidative stress induced by DUX4 is a direct cause of DNA damage, and both DNA damage and oxidative stress can also affect the myogenic differentiation process contributing to aberrant myotube formation [66]. Noteworthy is that DNA damage caused by moderate doses of oxidants is efficiently repaired in FSHD myoblasts, suggesting the capacity of handling oxidative stress up to a certain level [67]. Furthermore, DUX4 can activate TNF-α and JNK pathways, increasing susceptibility to oxidative stress-induced cell death, as well as regulate hypoxia-inducible factor 1α (HIF1A) expression, which, upon interacting with β-catenin, inhibits cell proliferation and induces transcription of hypoxic response genes [68].
DUX4 is also able to modulate the Nrf2-mediated oxidative stress response pathway. Sharma and colleagues, in a transcriptomic study conducted on human rhabdomyosarcoma cells overexpressing DUX4, identified 31 out of the 86 transcripts known to function in the Nrf2 pathway as differentially expressed, with the majority of these changes potentially inducing or contributing to oxidative stress [69]. The study confirms the link between Nrf2 and DUX4, and it further highlights the role of DUX4 in the induction of oxidative stress.
DUX4 seems to be negatively involved in the plasma membrane repair pathways as well. Indeed, Bittel and colleagues demonstrated, using a cellular and animal model, that DUX4 inhibition, as well as antioxidant treatment, is able to improve plasma membrane repair by reducing mitochondrial ROS levels [70].
Further evidence coming from earlier studies supports additional roles for oxidative stress in the development of muscle wasting in FSHD. They also showed an alteration of several genes involved in oxidative stress, particularly a glutathione S-transferase theta-2 (GSTT2) downregulation, suggesting a reduced capacity to buffer oxidative stress [71]. These structural alterations are associated with mitochondrial dysfunctions, such as reduced COX activity and ATP production, as well as oxidative stress imbalance [72]. PGC1α, which is critical for mitochondria biogenesis, is directly involved in defense against oxidative stress, upregulating different antioxidant enzymes such as Manganese superoxide dismutase (MnSOD).
It is intriguing that inflammation seems to be involved in the development of muscle damage in FSHD with a progression that can be followed using muscle imaging [73][74]. Although its role is not completely understood, in vivo evidence suggests that it constitutes an active process in muscles undergoing early damage [75] and significantly involves cytokines and mediators of the innate immunity arm [76]. The possible interplay between inflammation and oxidative stress in FSHD also needs clarification, as preliminary evidence suggests a redox imbalance in muscles showing signs of early damage. In these muscles, proteomic analysis of the interstitial fluid analyzed by mass spectrometry identified a downregulation of SOD1 and upregulation of GPx3 and CAT [77].
Oxidative stress is also a player in the pathophysiology of myotonic dystrophy type 1 (DM1). In 1995, Ihara and colleagues proposed a role for oxidative stress in DM1 on the basis of the discovery in patients’ blood of an increased level of free radicals and lipid peroxides, as well as a decrease in antioxidants such as α-tocopherol, coenzyme Q10, selenium, and albumin [78]. Usaki and colleagues, using the C2C12 cell model, demonstrated that the susceptibility to oxidative stress is CTG-repeat number-dependent, suggesting that it could be involved in the pathogenesis of the disease. Furthermore, using the same model, they found that the induction of apoptosis by oxidative stress is related to the activation of the SAPK/JNK pathway and the inhibition of the ERK pathway [79].
Moreover, evidence indicates that mitochondrial dysfunction is involved in the pathophysiology of DM1. In this regard, signs of mitochondrial alteration in muscle biopsy and oxidative stress markers have been detected in different DM1 patient cohorts [80][81]. Lastly, recent studies focused the attention on antioxidant system deregulation showing the downregulation of several proteins such as GPx, Gst, and GSH in DM1 patients [82][83].
Limb girdle muscular dystrophies (LGMDs) constitute a group of genetic disorders characterized by progressive weakness and wasting of the proximal limb muscles, with onset by definition after the acquisition of autonomous ambulation. Several studies showed the involvement of oxidative stress in calpainopathy (LGMDR1, previously named LGMD2A). Capn3 KO mice show abnormalities in mitochondrial structure, distribution, and function, suggesting that energy production deficits, along with increased oxidative stress, are pathogenic features of LGMDR1 [84]. Moreover, oxidative and nitrosative stress occurring in LGMDR1 activate different pathways such as the NF-κB
Redox imbalance has also been detected in LGMDR1 patients [85], where reductions in antioxidant defense mechanisms (SOD1 and Nrf2), coupled with increased lipid peroxidation and protein ubiquitination, were found. The redox imbalance primarily affected nonmitochondrial compartments, since the enzyme activities of citrate synthase, cytochrome c oxidase, and complex I + III were comparable to controls.
Lastly, a relevant upregulation of oxidative stress and NF-κB signaling has also been identified in dysferlinopathy or LGMDR2. Analyses on human precursor and differentiated cultured muscle cells demonstrated that a reduction in dysferlin induces oxidative stress with the mitochondria as a source of ROS [86].
5. Potential Targets for Therapy
As oxidative stress is involved in muscle homeostasis and functionality, antioxidant treatment may be a potential therapeutic option, alone or as an adjuvant, to treat myopathies and muscular dystrophies. To date, different studies have shown the ability of antioxidants to improve muscle health, reducing ROS levels through the modulation of different genes involved in oxidative stress response (Figure 2).
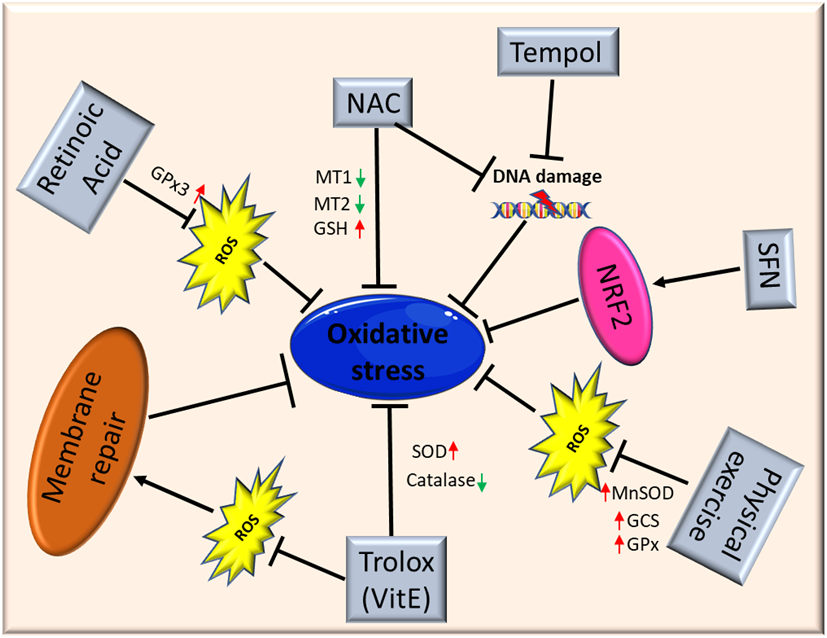
Figure 2. Schematic illustration of antioxidant therapy targets in muscular dystrophies. Abbreviations: ROS: Reactive Oxygen Species; NAC: N-Acetylcysteine; MT1: metallothionein1; MT2: metallothionein2; GSH: glutathione; SFN: isothiocyanate Sulforaphane; NFR2: Nuclear factor erythroid 2-related factor 2; MnSOD: Manganese superoxide dismutase; GCS: γ-glutamylcysteine synthetase; GPx: glutathione peroxidase; SOD: superoxide dismutase.
El Haddad and colleagues reported that treatment with retinoic acid, a metabolite of vitamin A, reduced ROS levels in human myoblasts derived from both healthy subjects and FSHD patients and improved cell survival in transplantation assays. Moreover, they underlined the molecular mechanism involved, further demonstrating that retinoic acid is able to induce expression and increase the activity of GPx3 [87]. Another study demonstrated that treatment with tempol, a powerful antioxidant, of DUX4-transfected myoblasts, as well as myoblasts derived from FSHD patients, efficiently reduced the level of ROS and DNA breaks. Similar findings were obtained using NAC[80]. Moreover, deferoxamine (DFX) was able to improve the antioxidant effects of NAC on primary cultures from mdx mice mainly reducing H2O2 production and NF-_B levels[88]. NAC treatment also decreased oxidative stress-responsive genes metallothionein1 and 2 (Mt1 and Mt2), ameliorating myopathic phenotypes in a mouse model of GNE myopathy, one of the most prevalent distalmyopathies[89]. Vitamin E is commonly used as an antioxidant to limit oxidative damage and inflammatory disease. Bittel and colleagues reported that treatment with Trolox, a water-soluble analogue of vitamin E, significantly improved membrane repair capacity by reducing mitochondrial ROS levels in FSHD myoblasts[70]. Furthermore, vitamin E is able to significantly increase the level of SOD and decrease the catalase level in mdx mice, suggesting that it may contribute to counteract the deleterious effects of oxidative damage, including lipid peroxidation and free-radical generation[56]. Bosnakovski and colleagues identified 52 compounds that can inhibit DUX4-induced toxicity in FSHD myoblasts. Two-thirds of these compounds have been shown to reduce oxidative stress in FSHD muscle, along with slowing disease processes[65]. Several studies also highlighted the role of physical exercise to counteract oxidative stress in muscle. Increasing evidence suggests that the continued presence of a small stimulus, such as low concentrations of ROS, can induce a compensatory increase of antioxidant defenses. It is important to underline once again that Nrf2 is the primary regulator of the endogenous antioxidant response, and that Nrf2 induction is a more efficient alternative to the use of a single antioxidant. Indeed, unlike other strategies, targeting Nrf2 activation has the potential to simultaneously modulate separate pathological features and amplify therapeutic benefits in muscular dystrophies[2]. Nrf2 may ensure a durable benefit in chronic disease states via upstream regulation of several antioxidant and anti-inflammatory pathways. Many Nrf2 activators have been identified, some of those are either already in clinical practice or tested in interventional trials (e.g., the fumaric acid esters, oltipraz and ursodiol)[90][91][92]. For instance, the use of the isothiocyanate sulforaphane (SFN), a well-known Nrf2 inducer[93], was able to attenuate muscle inflammation and fibrosis in mdx mice[94]. A recent study further confirmed the importance of targeting Nrf2 to amplify the therapeutic benefits and open the way to new drugs for treating chronic muscle diseases[2].
6. Conclusions
Evidence coming from the literature suggests that oxidative stress is an important modulator of skeletal muscle homeostasis and functionality. As discussed, oxidative stress is one of the most relevant causes of muscle damage, and it is not surprising that an impairment of redox homeostasis has been evidenced in several muscle disorders. Since redox homeostasis is drug-targetable, the implementation of pharmacological trials with antioxidants can pave the way for novel promising preclinical studies. Nevertheless, many questions need further investigation, such as the exact mechanism underlying oxidative stress in patients with muscle disorders and the effective benefits of antioxidant supplementation in specific forms of muscular dystrophies.
References
- Le Moal, E.; Pialoux, V.; Juban, G.; Groussard, C.; Zouhal, H.; Chazaud, B.; Mounier, R. Redox Control of Skeletal Muscle Regeneration. Antioxid. Redox Signal. 2017, 27, 276–310.
- Kourakis, S.; Timpani, C.A.; de Haan, J.B.; Gueven, N.; Fischer, D.; Rybalka, E. Targeting Nrf2 for the Treatment of Duchenne Muscular Dystrophy. Redox Biol. 2021, 38, 101803.
- Rojo de la Vega, M.; Dodson, M.; Chapman, E.; Zhang, D.D. NRF2-Targeted Therapeutics: New Targets and Modes of NRF2 Regulation. Curr. Opin. Toxicol. 2016, 1, 62–70.
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203.
- Serra, A.J.; Pinto, J.R.; Prokić, M.D.; Arsa, G.; Vasconsuelo, A. Oxidative Stress in Muscle Diseases: Current and Future Therapy. Oxid. Med. Cell. Longev. 2020, 2020, 6030417.
- Barbieri, E.; Sestili, P. Reactive Oxygen Species in Skeletal Muscle Signaling. J. Signal Transduct. 2012, 2012, 982794.
- Ghosh, S.; Karin, M. Missing Pieces in the NF-ΚB Puzzle. Cell 2002, 109, S81–S96.
- Jiang, B.; Xu, S.; Hou, X.; Pimentel, D.R.; Brecher, P.; Cohen, R.A. Temporal Control of NF-ΚB Activation by ERK Differentially Regulates Interleukin-1β-Induced Gene Expression. J. Biol. Chem. 2004, 279, 1323–1329.
- Li, Q.; Engelhardt, J.F. Interleukin-1β Induction of NFκB Is Partially Regulated by H2O2-Mediated Activation of NFκB-Inducing Kinase. J. Biol. Chem. 2006, 281, 1495–1505.
- Aoi, W.; Naito, Y.; Takanami, Y.; Kawai, Y.; Sakuma, K.; Ichikawa, H.; Yoshida, N.; Yoshikawa, T. Oxidative Stress and Delayed-Onset Muscle Damage after Exercise. Free Radic. Biol. Med. 2004, 37, 480–487.
- Hanai, J.; Cao, P.; Tanksale, P.; Imamura, S.; Koshimizu, E.; Zhao, J.; Kishi, S.; Yamashita, M.; Phillips, P.S.; Sukhatme, V.P.; et al. The Muscle-Specific Ubiquitin Ligase Atrogin-1/MAFbx Mediates Statin-Induced Muscle Toxicity. J. Clin. Investig. 2007, 117, 3940–3951.
- Sun, C.-C.; Li, S.-J.; Yang, C.-L.; Xue, R.-L.; Xi, Y.-Y.; Wang, L.; Zhao, Q.-L.; Li, D.-J. Sulforaphane Attenuates Muscle Inflammation in Dystrophin-Deficient Mdx Mice via NF-E2-Related Factor 2 (Nrf2)-Mediated Inhibition of NF-ΚB Signaling Pathway. J. Biol. Chem. 2015, 290, 17784–17795.
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 Talks, Who’s Listening? Antioxid. Redox Signal. 2010, 13, 1649–1663.
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 Signaling Pathway: Pivotal Roles in Inflammation. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 585–597.
- Seldon, M.P.; Silva, G.; Pejanovic, N.; Larsen, R.; Gregoire, I.P.; Filipe, J.; Anrather, J.; Soares, M.P. Heme Oxygenase-1 Inhibits the Expression of Adhesion Molecules Associated with Endothelial Cell Activation via Inhibition of NF-ΚB RelA Phosphorylation at Serine. J. Immunol. 2007, 179, 7840–7851.
- Wang, C.-H.; Chou, P.-C.; Chung, F.-T.; Lin, H.-C.; Huang, K.-H.; Kuo, H.-P. Heat Shock Protein70 Is Implicated in Modulating NF-ΚB Activation in Alveolar Macrophages of Patients with Active Pulmonary Tuberculosis. Sci. Rep. 2017, 7, 1214.
- Pietraszek-Gremplewicz, K.; Kozakowska, M.; Bronisz-Budzynska, I.; Ciesla, M.; Mucha, O.; Podkalicka, P.; Madej, M.; Glowniak, U.; Szade, K.; Stępniewski, J.; et al. Heme Oxygenase-1 Influences Satellite Cells and Progression of Duchenne Muscular Dystrophy in Mice. Antioxid. Redox Signal. 2018, 29, 128–148.
- Chan, M.C.; Ziegler, O.; Liu, L.; Rowe, G.C.; Das, S.; Otterbein, L.E.; Arany, Z. Heme Oxygenase and Carbon Monoxide Protect from Muscle Dystrophy. Skelet. Muscle 2016, 6, 41.
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 11624.
- Gilbreath, H.R.; Castro, D.; Iannaccone, S.T. Congenital Myopathies and Muscular Dystrophies. Neurol. Clin. 2014, 32, 689–703.
- Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Diagnostic Approach to the Congenital Muscular Dystrophies. Neuromuscul. Disord. NMD 2014, 24, 289–311.
- Canton, M.; Menazza, S.; Di Lisa, F. Oxidative Stress in Muscular Dystrophy: From Generic Evidence to Specific Sources and Targets. J. Muscle Res. Cell Motil. 2014, 35, 23–36.
- Terrill, J.R.; Radley-Crabb, H.G.; Iwasaki, T.; Lemckert, F.A.; Arthur, P.G.; Grounds, M.D. Oxidative Stress and Pathology in Muscular Dystrophies: Focus on Protein Thiol Oxidation and Dysferlinopathies. FEBS J. 2013, 280, 4149–4164.
- Kozakowska, M.; Pietraszek-Gremplewicz, K.; Jozkowicz, A.; Dulak, J. The Role of Oxidative Stress in Skeletal Muscle Injury and Regeneration: Focus on Antioxidant Enzymes. J. Muscle Res. Cell Motil. 2015, 36, 377–393.
- Choi, M.H.; Ow, J.R.; Yang, N.-D.; Taneja, R. Oxidative Stress-Mediated Skeletal Muscle Degeneration: Molecules, Mechanisms, and Therapies. Oxid. Med. Cell. Longev. 2016, 2016, 6842568.
- Michelucci, A.; De Marco, A.; Guarnier, F.A.; Protasi, F.; Boncompagni, S. Antioxidant Treatment Reduces Formation of Structural Cores and Improves Muscle Function in RYR1Y522S/WT Mice. Oxid. Med. Cell. Longev. 2017, 2017, 6792694.
- Arbogast, S.; Ferreiro, A. Selenoproteins and Protection against Oxidative Stress: Selenoprotein N as a Novel Player at the Crossroads of Redox Signaling and Calcium Homeostasis. Antioxid. Redox Signal. 2010, 12, 893–904.
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne Muscular Dystrophy: Myonecrosis, Inflammation and Oxidative Stress. Dis. Model. Mech. 2020, 13.
- Can, B.; Kara, O.; Kizilarslanoglu, M.C.; Arik, G.; Aycicek, G.S.; Sumer, F.; Civelek, R.; Demirtas, C.; Ulger, Z. Serum Markers of Inflammation and Oxidative Stress in Sarcopenia. Aging Clin. Exp. Res. 2017, 29, 745–752.
- Morrison, L. Unraveling RYR1 Mutations and Muscle Biopsies. Neurology 2008, 70, 99–100.
- Snoeck, M.; van Engelen, B.G.M.; Küsters, B.; Lammens, M.; Meijer, R.; Molenaar, J.P.F.; Raaphorst, J.; Verschuuren-Bemelmans, C.C.; Straathof, C.S.M.; Sie, L.T.L.; et al. RYR1-Related Myopathies: A Wide Spectrum of Phenotypes throughout Life. Eur. J. Neurol. 2015, 22, 1094–1112.
- Lopez, R.J.; Byrne, S.; Vukcevic, M.; Sekulic-Jablanovic, M.; Xu, L.; Brink, M.; Alamelu, J.; Voermans, N.; Snoeck, M.; Clement, E.; et al. An RYR1 Mutation Associated with Malignant Hyperthermia Is Also Associated with Bleeding Abnormalities. Sci. Signal. 2016, 9, ra68.
- Treves, S.; Jungbluth, H.; Muntoni, F.; Zorzato, F. Congenital Muscle Disorders with Cores: The Ryanodine Receptor Calcium Channel Paradigm. Curr. Opin. Pharmacol. 2008, 8, 319–326.
- Meissner, G. The Structural Basis of Ryanodine Receptor Ion Channel Function. J. Gen. Physiol. 2017, 149, 1065–1089.
- Hamilton, S.L.; Reid, M.B. RyR1 Modulation by Oxidation and Calmodulin. Antioxid. Redox Signal. 2000, 2, 41–45.
- Aracena-Parks, P.; Goonasekera, S.A.; Gilman, C.P.; Dirksen, R.T.; Hidalgo, C.; Hamilton, S.L. Identification of Cysteines Involved in S-Nitrosylation, S-Glutathionylation, and Oxidation to Disulfides in Ryanodine Receptor Type. J. Biol. Chem. 2006, 281, 40354–40368.
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine Receptor Oxidation Causes Intracellular Calcium Leak and Muscle Weakness in Aging. Cell Metab. 2011, 14, 196–207.
- Durham, W.J.; Aracena-Parks, P.; Long, C.; Rossi, A.E.; Goonasekera, S.A.; Boncompagni, S.; Galvan, D.L.; Gilman, C.P.; Baker, M.R.; Shirokova, N.; et al. RyR1 S-Nitrosylation Underlies Environmental Heat Stroke and Sudden Death in Y522S RyR1 Knockin Mice. Cell 2008, 133, 53–65.
- Todd, J.J.; Lawal, T.A.; Witherspoon, J.W.; Chrismer, I.C.; Razaqyar, M.S.; Punjabi, M.; Elliott, J.S.; Tounkara, F.; Kuo, A.; Shelton, M.O.; et al. Randomized Controlled Trial of N-Acetylcysteine Therapy for RYR1-Related Myopathies. Neurology 2020, 94, e1434–e1444.
- Dowling, J.J.; Arbogast, S.; Hur, J.; Nelson, D.D.; McEvoy, A.; Waugh, T.; Marty, I.; Lunardi, J.; Brooks, S.V.; Kuwada, J.Y.; et al. Oxidative Stress and Successful Antioxidant Treatment in Models of RYR1-Related Myopathy. Brain J. Neurol. 2012, 135, 1115–1127.
- Lee, C.S.; Hanna, A.D.; Wang, H.; Dagnino-Acosta, A.; Joshi, A.D.; Knoblauch, M.; Xia, Y.; Georgiou, D.K.; Xu, J.; Long, C.; et al. A Chemical Chaperone Improves Muscle Function in Mice with a RyR1 Mutation. Nat. Commun. 2017, 8, 14659.
- Flanigan, K.M.; Kerr, L.; Bromberg, M.B.; Leonard, C.; Tsuruda, J.; Zhang, P.; Gonzalez-Gomez, I.; Cohn, R.; Campbell, K.P.; Leppert, M. Congenital Muscular Dystrophy with Rigid Spine Syndrome: A Clinical, Pathological, Radiological, and Genetic Study. Ann. Neurol. 2000, 47, 152–161.
- Rederstorff, M.; Castets, P.; Arbogast, S.; Lainé, J.; Vassilopoulos, S.; Beuvin, M.; Dubourg, O.; Vignaud, A.; Ferry, A.; Krol, A.; et al. Increased Muscle Stress-Sensitivity Induced by Selenoprotein N Inactivation in Mouse: A Mammalian Model for SEPN1-Related Myopathy. PLoS ONE 2011, 6, e23094.
- Arbogast, S.; Beuvin, M.; Fraysse, B.; Zhou, H.; Muntoni, F.; Ferreiro, A. Oxidative Stress in SEPN1-Related Myopathy: From Pathophysiology to Treatment. Ann. Neurol. 2009, 65, 677–686.
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Pharmacological and Psychosocial Management. Lancet Neurol. 2010, 9, 77–93.
- Matsumura, K.; Campbell, K.P. Dystrophin-Glycoprotein Complex: Its Role in the Molecular Pathogenesis of Muscular Dystrophies. Muscle Nerve 1994, 17, 2–15.
- Serra, F.; Quarta, M.; Canato, M.; Toniolo, L.; De Arcangelis, V.; Trotta, A.; Spath, L.; Monaco, L.; Reggiani, C.; Naro, F. Inflammation in Muscular Dystrophy and the Beneficial Effects of Non-Steroidal Anti-Inflammatory Drugs. Muscle Nerve 2012, 46, 773–784.
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS Signaling: Rapid Mechano-Chemo Transduction in Heart. Science 2011, 333, 1440–1445.
- Rando, T.A.; Disatnik, M.H.; Yu, Y.; Franco, A. Muscle Cells from Mdx Mice Have an Increased Susceptibility to Oxidative Stress. Neuromuscul. Disord. NMD 1998, 8, 14–21.
- Niebrój-Dobosz, I.; Hausmanowa-Petrusewicz, I. The Involvement of Oxidative Stress in Determining the Severity and Progress of Pathological Processes in Dystrophin-Deficient Muscles. Acta Biochim. Pol. 2005, 52, 449–452.
- Kaczor, J.J.; Hall, J.E.; Payne, E.; Tarnopolsky, M.A. Low Intensity Training Decreases Markers of Oxidative Stress in Skeletal Muscle of Mdx Mice. Free Radic. Biol. Med. 2007, 43, 145–154.
- Kim, J.-H.; Kwak, H.-B.; Thompson, L.V.; Lawler, J.M. Contribution of Oxidative Stress to Pathology in Diaphragm and Limb Muscles with Duchenne Muscular Dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 1–13.
- Pal, R.; Palmieri, M.; Loehr, J.A.; Li, S.; Abo-Zahrah, R.; Monroe, T.O.; Thakur, P.B.; Sardiello, M.; Rodney, G.G. Src-Dependent Impairment of Autophagy by Oxidative Stress in a Mouse Model of Duchenne Muscular Dystrophy. Nat. Commun. 2014, 5, 4425.
- Petrillo, S.; Pelosi, L.; Piemonte, F.; Travaglini, L.; Forcina, L.; Catteruccia, M.; Petrini, S.; Verardo, M.; D’Amico, A.; Musarò, A.; et al. Oxidative Stress in Duchenne Muscular Dystrophy: Focus on the NRF2 Redox Pathway. Hum. Mol. Genet. 2017, 26, 2781–2790.
- Almeida-Becerril, T.; Rodríguez-Cruz, M.; Raúl Sánchez-González, J.; Antonio Villaldama-Soriano, M.; Atilano-Miguel, S.; Villa-Morales, J.; Cárdenas-Conejo, A.; Cárdenas-Vázquez, R. Circulating Markers of Oxidative Stress Are Associated with a Muscle Injury in Patients with Muscular Dystrophy Duchenne. Brain Dev. 2021, 43, 111–120.
- Mâncio, R.D.; Hermes, T.d.A.; Macedo, A.B.; Mizobuti, D.S.; Valduga, A.H.; Rupcic, I.F.; Minatel, E. Vitamin E Treatment Decreases Muscle Injury in Mdx Mice. Nutrition 2017, 43–44, 39–46.
- Mizobuti, D.S.; Fogaça, A.R.; Moraes, F.D.S.R.; Moraes, L.H.R.; Mâncio, R.D.; Hermes, T.d.A.; Macedo, A.B.; Valduga, A.H.; de Lourenço, C.C.; Pereira, E.C.L.; et al. Coenzyme Q10 Supplementation Acts as Antioxidant on Dystrophic Muscle Cells. Cell Stress Chaperones 2019, 24, 1175–1185.
- Burns, D.P.; Drummond, S.E.; Bolger, D.; Coiscaud, A.; Murphy, K.H.; Edge, D.; O’Halloran, K.D. N-Acetylcysteine Decreases Fibrosis and Increases Force-Generating Capacity of Mdx Diaphragm. Antioxidants 2019, 8, 581.
- Moraes, L.H.R.; Bollineli, R.C.; Mizobuti, D.S.; Silveira, L.D.R.; Marques, M.J.; Minatel, E. Effect of N-Acetylcysteine plus Deferoxamine on Oxidative Stress and Inflammation in Dystrophic Muscle Cells. Redox Rep. Commun. Free Radic. Res. 2015, 20, 109–115.
- Pinniger, G.J.; Terrill, J.R.; Assan, E.B.; Grounds, M.D.; Arthur, P.G. Pre-Clinical Evaluation of N-Acetylcysteine Reveals Side Effects in the Mdx Mouse Model of Duchenne Muscular Dystrophy. J. Physiol. 2017, 595, 7093–7107.
- Gamberi, T.; Fiaschi, T.; Valocchia, E.; Modesti, A.; Mantuano, P.; Rolland, J.-F.; Sanarica, F.; De Luca, A.; Magherini, F. Proteome Analysis in Dystrophic Mdx Mouse Muscle Reveals a Drastic Alteration of Key Metabolic and Contractile Proteins after Chronic Exercise and the Potential Modulation by Anti-Oxidant Compounds. J. Proteomics 2018, 170, 43–58.
- Lemmers, R.J.L.F.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy. Science 2010, 329, 1650–1653.
- Hamel, J.; Tawil, R. Facioscapulohumeral Muscular Dystrophy: Update on Pathogenesis and Future Treatments. Neurother. J. Am. Soc. Exp. Neurother. 2018, 15, 863–871.
- Bosnakovski, D.; Xu, Z.; Ji Gang, E.; Galindo, C.L.; Liu, M.; Simsek, T.; Garner, H.R.; Agha-Mohammadi, S.; Tassin, A.; Coppée, F.; et al. An Isogenetic Myoblast Expression Screen Identifies DUX4-Mediated FSHD-Associated Molecular Pathologies. EMBO J. 2008, 27, 2766–2779.
- Bosnakovski, D.; Choi, S.; Strasser, J.M.; Toso, E.A.; Walters, M.A.; Kyba, M. High-Throughput Screening Identifies Inhibitors of DUX4-Induced Myoblast Toxicity. Skelet. Muscle 2014, 4, 4.
- Dmitriev, P.; Bou Saada, Y.; Dib, C.; Ansseau, E.; Barat, A.; Hamade, A.; Dessen, P.; Robert, T.; Lazar, V.; Louzada, R.A.N.; et al. DUX4-Induced Constitutive DNA Damage and Oxidative Stress Contribute to Aberrant Differentiation of Myoblasts from FSHD Patients. Free Radic. Biol. Med. 2016, 99, 244–258.
- Bou Saada, Y.; Dib, C.; Dmitriev, P.; Hamade, A.; Carnac, G.; Laoudj-Chenivesse, D.; Lipinski, M.; Vassetzky, Y.S. Facioscapulohumeral Dystrophy Myoblasts Efficiently Repair Moderate Levels of Oxidative DNA Damage. Histochem. Cell Biol. 2016, 145, 475–483.
- Banerji, C.R.S.; Knopp, P.; Moyle, L.A.; Severini, S.; Orrell, R.W.; Teschendorff, A.E.; Zammit, P.S. β– Catenin Is Central to DUX4 -Driven Network Rewiring in Facioscapulohumeral Muscular Dystrophy. J. R. Soc. Interface 2015, 12, 20140797.
- Sharma, V.; Harafuji, N.; Belayew, A.; Chen, Y.-W. DUX4 Differentially Regulates Transcriptomes of Human Rhabdomyosarcoma and Mouse C2C12 Cells. PLoS ONE 2013, 8, e64691.
- Bittel, A.J.; Sreetama, S.C.; Bittel, D.C.; Horn, A.; Novak, J.S.; Yokota, T.; Zhang, A.; Maruyama, R.; Rowel, Q.; Lim, K.; et al. Membrane Repair Deficit in Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 5575.
- Winokur, S.T.; Barrett, K.; Martin, J.H.; Forrester, J.R.; Simon, M.; Tawil, R.; Chung, S.-A.; Masny, P.S.; Figlewicz, D.A. Facioscapulohumeral Muscular Dystrophy (FSHD) Myoblasts Demonstrate Increased Susceptibility to Oxidative Stress. Neuromuscul. Disord. 2003, 13, 322–333.
- Turki, A.; Hayot, M.; Carnac, G.; Pillard, F.; Passerieux, E.; Bommart, S.; de Mauverger, E.R.; Hugon, G.; Pincemail, J.; Pietri, S.; et al. Functional Muscle Impairment in Facioscapulohumeral Muscular Dystrophy Is Correlated with Oxidative Stress and Mitochondrial Dysfunction. Free Radic. Biol. Med. 2012, 53, 1068–1079.
- Frisullo, G.; Frusciante, R.; Nociti, V.; Tasca, G.; Renna, R.; Iorio, R.; Patanella, A.K.; Iannaccone, E.; Marti, A.; Rossi, M.; et al. CD8(+) T Cells in Facioscapulohumeral Muscular Dystrophy Patients with Inflammatory Features at Muscle MRI. J. Clin. Immunol. 2011, 31, 155–166.
- Monforte, M.; Laschena, F.; Ottaviani, P.; Bagnato, M.R.; Pichiecchio, A.; Tasca, G.; Ricci, E. Tracking Muscle Wasting and Disease Activity in Facioscapulohumeral Muscular Dystrophy by Qualitative Longitudinal Imaging. J. Cachexia Sarcopenia Muscle 2019, 10, 1258–1265.
- Tasca, G.; Monforte, M.; Iannaccone, E.; Laschena, F.; Ottaviani, P.; Leoncini, E.; Boccia, S.; Galluzzi, G.; Pelliccioni, M.; Masciullo, M.; et al. Upper Girdle Imaging in Facioscapulohumeral Muscular Dystrophy. PLoS ONE 2014, 9, e100292.
- Tasca, G.; Monforte, M.; Corbi, M.; Granata, G.; Lucchetti, D.; Sgambato, A.; Ricci, E. Muscle Microdialysis to Investigate Inflammatory Biomarkers in Facioscapulohumeral Muscular Dystrophy. Mol. Neurobiol. 2018, 55, 2959–2966.
- Corasolla Carregari, V.; Monforte, M.; Di Maio, G.; Pieroni, L.; Urbani, A.; Ricci, E.; Tasca, G. Proteomics of Muscle Microdialysates Identifies Potential Circulating Biomarkers in Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 22, 290.
- Ihara, Y.; Mori, A.; Hayabara, T.; Namba, R.; Nobukuni, K.; Sato, K.; Miyata, S.; Edamatsu, R.; Liu, J.; Kawai, M. Free Radicals, Lipid Peroxides and Antioxidants in Blood of Patients with Myotonic Dystrophy. J. Neurol. 1995, 242, 119–122.
- Usuki, F.; Takahashi, N.; Sasagawa, N.; Ishiura, S. Differential Signaling Pathways Following Oxidative Stress in Mutant Myotonin Protein Kinase CDNA-Transfected C2C12 Cell Lines. Biochem. Biophys. Res. Commun. 2000, 267, 739–743.
- Toscano, A.; Messina, S.; Campo, G.M.; Leo, R.D.; Rodolico, C.; Aguennouz, M.; Annesi, G.; Vita, G. Oxidative Stress in Myotonic Dystrophy Type 1. Free Radic. Res. 2005, 39, 771–776.
- Gramegna, L.L.; Giannoccaro, M.P.; Manners, D.N.; Testa, C.; Zanigni, S.; Evangelisti, S.; Bianchini, C.; Oppi, F.; Poda, R.; Avoni, P.; et al. Mitochondrial Dysfunction in Myotonic Dystrophy Type. Neuromuscul. Disord. 2018, 28, 144–149.
- Kumar, A.; Kumar, V.; Singh, S.K.; Muthuswamy, S.; Agarwal, S. Imbalanced Oxidant and Antioxidant Ratio in Myotonic Dystrophy Type. Free Radic. Res. 2014, 48, 503–510.
- Koc, F.; Atli, G.; Menziletoglu, S.Y.; Kose, S. Antioxidant Imbalance in the Erythrocytes of Myotonic Dystrophy Type 1 Patients. Arch. Biochem. Biophys. 2020, 680, 108230.
- Kramerova, I.; Kudryashova, E.; Wu, B.; Germain, S.; Vandenborne, K.; Romain, N.; Haller, R.G.; Verity, M.A.; Spencer, M.J. Mitochondrial Abnormalities, Energy Deficit and Oxidative Stress Are Features of Calpain 3 Deficiency in Skeletal Muscle. Hum. Mol. Genet. 2009, 18, 3194–3205.
- Nilsson, M.I.; Macneil, L.G.; Kitaoka, Y.; Alqarni, F.; Suri, R.; Akhtar, M.; Haikalis, M.E.; Dhaliwal, P.; Saeed, M.; Tarnopolsky, M.A. Redox State and Mitochondrial Respiratory Chain Function in Skeletal Muscle of LGMD2A Patients. PLoS ONE 2014, 9, e102549.
- Rajakumar, D.; Senguttuvan, S.; Alexander, M.; Oommen, A. Involvement of Oxidative Stress, Nuclear Factor Kappa B and the Ubiquitin Proteasomal Pathway in Dysferlinopathy. Life Sci. 2014, 8, 54–61.
- Marina El Haddad; Elise Jean; Ahmed Turki; Gérald Hugon; Barbara Vernus; Anne Bonnieu; Emilie Passerieux; Aline Hamade; Jacques Mercier; Dalila Laoudj-Chenivesse; et al.Gilles Carnac Glutathione peroxidase 3, a new retinoid target gene, is critical for human skeletal muscle precursor cell survival. Journal of Cell Science 2012, 125, 6147-6156, 10.1242/jcs.115220.
- Luis Henrique Rapucci Moraes; Roberta Constâncio Bollineli; Daniela Sayuri Mizobuti; Leonardo Dos Reis Silveira; Maria Julia Marques; Elaine Minatel; Effect of N-acetylcysteine plus deferoxamine on oxidative stress and inflammation in dystrophic muscle cells. Redox Report 2014, 20, 109-115, 10.1179/1351000214Y.0000000112.
- Anna Cho; May Christine; V. Malicdan; Miho Miyakawa; Ikuya Nonaka; Ichizo Nishino; Satoru Noguchi; Sialic acid deficiency is associated with oxidative stress leading to muscle atrophy and weakness in GNE myopathy. Human Molecular Genetics 2017, 26, 3081-3093, 10.1093/hmg/ddx192.
- Sarah A. Scuderi; Alessio Ardizzone; Irene Paterniti; Emanuela Esposito; Michela Campolo; Antioxidant and Anti-Inflammatory Effect of Nrf2 Inducer Dimethyl Fumarate in Neurodegenerative Diseases. Antioxidants 2020, 9, 630, 10.3390/antiox9070630.
- Danica Michaličková; Tomas Hrncir; Nikolina Kutinová Canová; Ondřej Slanař; Targeting Keap1/Nrf2/ARE signaling pathway in multiple sclerosis. European Journal of Pharmacology 2020, 873, 172973, 10.1016/j.ejphar.2020.172973.
- Natalia Robledinos-Antón; Raquel Fernández-Ginés; Gina Manda; Antonio Cuadrado; Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxidative Medicine and Cellular Longevity 2019, 2019, 1-20, 10.1155/2019/9372182.
- Claudia Tonelli; Iok In Christine Chio; David A. Tuveson; Transcriptional Regulation by Nrf2. Antioxidants & Redox Signaling 2018, 29, 1727-1745, 10.1089/ars.2017.7342.
- Chengcao Sun; Shu-Jun Li; Cui-Li Yang; Rui-Lin Xue; Yong-Yong Xi; Liang Wang; Qian-Long Zhao; De-Jia Li; Sulforaphane Attenuates Muscle Inflammation in Dystrophin-deficient mdx Mice via NF-E2-related Factor 2 (Nrf2)-mediated Inhibition of NF-κB Signaling Pathway. Journal of Biological Chemistry 2015, 290, 17784-17795, 10.1074/jbc.m115.655019.


