 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | ALESSANDRA STASI | + 3340 word(s) | 3340 | 2021-06-07 10:41:25 | | | |
| 2 | Vivi Li | Meta information modification | 3340 | 2021-06-11 03:04:27 | | |
Video Upload Options
High-density lipoproteins (HDLs) are a class of blood particles, principally involved in mediating reverse cholesterol transport from peripheral tissue to liver. Omics approaches have identified crucial mediators in the HDL proteomic and lipidomic profile, which are involved in distinct pleiotropic functions. Besides their role as cholesterol transporter, HDLs display anti-inflammatory, anti-apoptotic, anti-thrombotic, and anti-infection properties. Experimental and clinical studies have unveiled significant changes in both HDL serum amount and composition that lead to dysregulated host immune response and endothelial dysfunction in the course of sepsis. Most SARS-Coronavirus-2-infected patients admitted to the intensive care unit showed common features of sepsis disease, such as the overwhelmed systemic inflammatory response and the alterations in serum lipid profile. Despite relevant advances, episodes of mild to moderate acute kidney injury (AKI), occurring during systemic inflammatory diseases, are associated with long-term complications, and high risk of mortality. The multi-faceted relationship of kidney dysfunction with dyslipidemia and inflammation encourages to deepen the clarification of the mechanisms connecting these elements.
1. Introduction
2. HDLs Composition and Reverse Cholesterol Transport Function
3. Kidney Involvement in Sepsis Disease: From HDL Target to Modulator
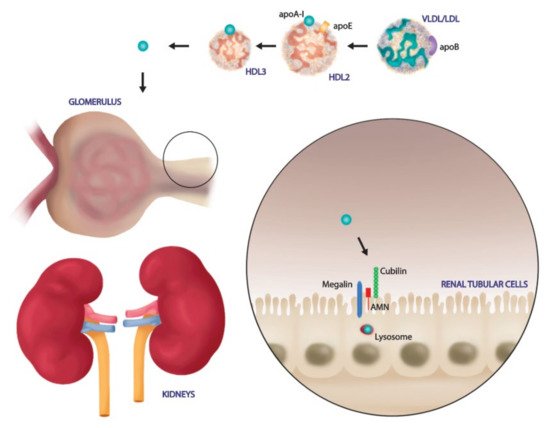
4. SARS-CoV-2 Exploits the SR-BI Associated HDL to Promote its Entry
References
- Khovidhunkit, W.; Kim, M.S.; Memon, R.A.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J. Lipid Res. 2004, 45, 1169–1196.
- Singh, I.M.; Shishehbor, M.H.; Ansell, B.J. High-density lipoprotein as a therapeutic target: A systematic review. J. Am. Med. Assoc. 2007, 298, 786–798.
- Navab, M.; Reddy, S.T.; Van Lenten, B.J.; Fogelman, A.M. HDL and cardiovascular disease: Atherogenic and atheroprotective mechanisms. Nat. Rev. Cardiol. 2011, 8, 222–232.
- Emancipator, K.; Csako, G.; Elin, R.J. In vitro inactivation of bacterial endotoxin by human lipoproteins and apolipoproteins. Infect. Immun. 1991, 60, 596–601.
- Munford, R.S. Detoxifying endotoxin: Time, place and person. J. Endotoxin Res. 2005, 11, 69–84.
- Lee, R.P.; Lin, N.T.; Chao, Y.F.C.; Lin, C.C.; Harn, H.J.; Chen, H.I. High-density lipoprotein prevents organ damage in endotoxemia. Res. Nurs. Health 2007, 30, 250–260.
- Grunfeld, C.; Marshall, M.; Shigenaga, J.K.; Moser, A.H.; Tobias, P.; Feingold, K.R. Lipoproteins inhibit macrophage activation by lipoteichoic acid. J. Lipid Res. 1999, 40, 245–252.
- Guo, L.; Ai, J.; Zheng, Z.; Howatt, D.A.; Daugherty, A.; Huang, B.; Li, X.-A. High density lipoprotein protects against polymicrobe-induced sepsis in mice. J. Biol. Chem. 2013, 288, 17947–17953.
- Guo, L.; Song, Z.; Li, M.; Wu, Q.; Wang, D.; Feng, H.; Bernard, P.; Daugherty, A.; Huang, B.; Li, X.A. Scavenger receptor BI protects against septic death through its role in modulating inflammatory response. J. Biol. Chem. 2009, 284, 19826–19834.
- Feingold, K.R.; Grunfeld, C. Lipids: A key player in the battle between the host and microorganisms. J. Lipid Res. 2012, 53, 2487–2489.
- van Leeuwen, H.J.; Heezius, E.C.J.M.; Dallinga, G.M.; van Strijp, J.A.G.; Verhoef, J.; van Kessel, K.P.M. Lipoprotein metabolism in patients with severe sepsis. Crit. Care Med. 2003, 31, 1359–1366.
- Chien, J.-Y.; Jerng, J.-S.; Yu, C.-J.; Yang, P.-C. Low serum level of high-density lipoprotein cholesterol is a poor prognostic factor for severe sepsis. Crit. Care Med. 2005, 33, 1688–1693.
- Tsai, M.-H.; Peng, Y.-S.; Chen, Y.-C.; Lien, J.-M.; Tian, Y.-C.; Fang, J.-T.; Weng, H.-H.; Chen, P.-C.; Yang, C.-W.; Wu, C.-S. Low serum concentration of apolipoprotein A-I is an indicator of poor prognosis in cirrhotic patients with severe sepsis. J. Hepatol. 2009, 50, 906–915.
- Eggesbø, J.B.; Hjermann, I.; Høstmark, A.T.; Kierulf, P. LPS induced release of IL-1β, IL-6, IL-8 and TNF-α in EDTA or heparin anticoagulated whole blood from persons with high or low levels of serum HDL. Cytokine 1996, 8, 152–160.
- Morin, E.E.; Guo, L.; Schwendeman, A.; Li, X.A. HDL in sepsis-risk factor and therapeutic approach. Front. Pharmacol. 2015, 6.
- Roveran Genga, K.; Lo, C.; Cirstea, M.; Zhou, G.; Walley, K.R.; Russell, J.A.; Levin, A.; Boyd, J.H. Two-year follow-up of patients with septic shock presenting with low HDL: The effect upon acute kidney injury, death and estimated glomerular filtration rate. J. Intern. Med. 2017, 281, 518–529.
- Zhang, Z.; Datta, G.; Zhang, Y.; Miller, A.P.; Mochon, P.; Chen, Y.F.; Chatham, J.; Anantharamaiah, G.M.; White, C.R. Apolipoprotein A-I mimetic peptide treatment inhibits inflammatory responses and improves survival in septic rats. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H866–H873.
- Yang, H.; Fogo, A.B.; Kon, V. Kidneys: Key modulators of high-density lipoprotein levels and function. Curr. Opin. Nephrol. Hypertens. 2016, 25, 174–179.
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, 1517–1520.
- Remy, K.E.; Brakenridge, S.C.; Francois, B.; Daix, T.; Deutschman, C.S.; Monneret, G.; Jeannet, R.; Laterre, P.F.; Hotchkiss, R.S.; Moldawer, L.L. Immunotherapies for COVID-19: Lessons learned from sepsis. Lancet Respir. Med. 2020, 8, 946–949.
- Sun, P.; Lu, X.; Xu, C.; Sun, W.; Pan, B. Understanding of COVID-19 based on current evidence. J. Med. Virol. 2020, 92, 548–551.
- Perico, L.; Benigni, A.; Remuzzi, G. Should COVID-19 Concern Nephrologists? Why and to What Extent? the Emerging Impasse of Angiotensin Blockade. Nephron 2020, 144, 213–221.
- World Health Organization. Mission China Joint Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (COVID-19); World Health Organization: Geneva, Switzerland, 2020; Volume 2019, pp. 16–24. Available online: (accessed on 7 April 2020).
- Feingold, K.R. Introduction to Lipids and Lipoproteins; MDText.com, Inc.: South Dartmouth, MA, USA, 2000.
- Otvos, J.D. Measurement of lipoprotein subclass profiles by nuclear magnetic resonance spectroscopy. Clin. Lab. 2002, 48, 171–180.
- Von Eckardstein, A.; Nofer, J.R.; Assmann, G. High density lipoproteins and arteriosclerosis role of cholesterol efflux and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 13–27.
- Suc, I.; Escargueil-Blanc, I.; Troly, M.; Salvayre, R.; Negre-Salvayre, A. HDL and apoA prevent cell death of endothelial cells induced by oxidized LDL. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2158–2166.
- Fielding, C.J.; Fielding, P.E. Molecular physiology of reverse cholesterol transport. J. Lipid Res. 1995, 36, 211–228.
- Lewis, G.F.; Rader, D.J. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res. 2005, 96, 1221–1232.
- Shah, A.S.; Tan, L.; Long, J.L.; Davidson, W.S. Proteomic diversity of high density lipoproteins: Our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 2013, 54, 2575–2585.
- Thompson, P.A.; Berbée, J.F.P.; Rensen, P.C.N.; Kitchens, R.L. Apolipoprotein A-II augments monocyte responses to LPS by suppressing the inhibitory activity of LPS-binding protein. Innate Immun. 2008, 14, 365–374.
- Furlaneto, C.J.; Ribeiro, F.P.; Hatanaka, E.; Souza, G.M.; Cassatella, M.A.; Campa, A. Apolipoproteins A-I and A-II downregulate neutrophil functions. Lipids 2002, 37, 925–928.
- Schuchardt, M.; Tölle, M.; Prüfer, J.; Van Der Giet, M. Pharmacological relevance and potential of sphingosine 1-phosphate in the vascular system. Br. J. Pharmacol. 2011, 163, 1140–1162.
- Kurano, M.; Tsuneyama, K.; Morimoto, Y.; Shimizu, T.; Jona, M.; Kassai, H.; Nakao, K.; Aiba, A.; Yatomi, Y. Apolipoprotein M Protects Lipopolysaccharide-Treated Mice from Death and Organ Injury. Thromb. Haemost. 2018, 118, 1021–1035.
- Karlsson, H.; Leanderson, P.; Tagesson, C.; Lindahl, M. Lipoproteomics I: Mapping of proteins in low-density lipoprotein using two-dimensional gel electrophoresis and mass spectrometry. Proteomics 2005, 5, 551–565.
- Moreno, J.A.; Ortega-Gomez, A.; Rubio-Navarro, A.; Louedec, L.; Ho-Tin-Noé, B.; Caligiuri, G.; Nicoletti, A.; Levoye, A.; Plantier, L.; Meilhac, O. High-density lipoproteins potentiate α1-antitrypsin therapy in elastase-induced pulmonary emphysema. Am. J. Respir. Cell Mol. Biol. 2014, 51, 536–549.
- Bricarello, D.A.; Mills, E.J.; Petrlova, J.; Voss, J.C.; Parikh, A.N. Ganglioside embedded in reconstituted lipoprotein binds cholera toxin with elevated affinity. J. Lipid Res. 2010, 51, 2731–2738.
- Okamoto, Y.; Wang, F.; Yoshioka, K.; Takuwa, N.; Takuwa, Y. Sphingosine-1-phosphate-specific G protein-coupled receptors as novel therapeutic targets for atherosclerosis. Pharmaceuticals 2011, 4, 117–137.
- Kimura, T.; Tomura, H.; Mogi, C.; Kuwabara, A.; Damirin, A.; Ishizuka, T.; Sekiguchi, A.; Ishiwara, M.; Im, D.S.; Sato, K.; et al. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells. J. Biol. Chem. 2006, 281, 37457–37467.
- Zhang, B.; Tomura, H.; Kuwabara, A.; Kimura, T.; Miura, S.I.; Noda, K.; Okajima, F.; Saku, K. Correlation of high density lipoprotein (HDL)-associated sphingosine 1-phosphate with serum levels of HDL-cholesterol and apolipoproteins. Atherosclerosis 2005, 178, 199–205.
- Tolle, M.; Pawlak, A.; Schuchardt, M.; Kawamura, A.; Tietge, U.J.; Lorkowski, S.; Keul, P.; Assmann, G.; Chun, J.; Levkau, B.; et al. HDL-associated lysosphingolipids inhibit NAD(P)H oxidase-dependent monocyte chemoattractant protein-1 production. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1542–1548.
- Winkler, M.S.; Nierhaus, A.; Holzmann, M.; Mudersbach, E.; Bauer, A.; Robbe, L.; Zahrte, C.; Geffken, M.; Peine, S.; Schwedhelm, E.; et al. Decreased serum concentrations of sphingosine-1-phosphate in sepsis. Crit. Care 2015, 19.
- Winkler, M.S.; Nierhaus, A.; Poppe, A.; Greiwe, G.; Gräler, M.H.; Daum, G. Sphingosine-1-Phosphate: A Potential Biomarker and Therapeutic Target for Endothelial Dysfunction and Sepsis? Shock 2017, 47, 666–672.
- Hoste, E.A.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423.
- Fiorentino, M.; Tohme, F.A.; Wang, S.; Murugan, R.; Angus, D.C.; Kellum, J.A. Long-term survival in patients with septic acute kidney injury is strongly influenced by renal recovery. PLoS ONE 2018, 13, e0198269.
- Seymour, C.W.; Liu, V.X.; Iwashyna, T.J.; Brunkhorst, F.M.; Rea, T.D.; Scherag, A.; Rubenfeld, G.; Kahn, J.M.; Shankar-Hari, M.; Singer, M.; et al. Assessment of Clinical Criteria for Sepsis: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 762–774.
- Bagshaw, S.M.; Uchino, S.; Bellomo, R.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; Gibney, N.; et al. Septic acute kidney injury in critically ill patients: Clinical characteristics and outcomes. Clin. J. Am. Soc. Nephrol. 2007, 2, 431–439.
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gómez, H.; Kellum, J.A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019, 96, 1083–1099.
- Kashani, K.; Al-Khafaji, A.; Ardiles, T.; Artigas, A.; Bagshaw, S.M.; Bell, M.; Bihorac, A.; Birkhahn, R.; Cely, C.M.; Chawla, L.S.; et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit. Care 2013, 17, R25.
- Fiorentino, M.; Xu, Z.; Smith, A.; Singbartl, K.; Palevsky, P.M.; Chawla, L.S.; Huang, D.T.; Yealy, D.M.; Angus, D.C.; Kellum, J.A. Serial Measurement of Cell-cycle Arrest Biomarkers [TIMP-2] [IGFBP7] and Risk for Progression to Death, Dialysis or Severe Acute Kidney Injury in Patients with Septic Shock. Am. J. Respir. Crit. Care Med. 2020, 202.
- Chawla, L.S.; Seneff, M.G.; Nelson, D.R.; Williams, M.; Levy, H.; Kimmel, P.L.; Macias, W.L. Elevated plasma concentrations of IL-6 and elevated APACHE II score predict acute kidney injury in patients with severe sepsis. Clin. J. Am. Soc. Nephrol. 2007, 2, 22–30.
- Cirstea, M.; Walley, K.R.; Russell, J.A.; Brunham, L.R.; Genga, K.R.; Boyd, J.H. Decreased high-density lipoprotein cholesterol level is an early prognostic marker for organ dysfunction and death in patients with suspected sepsis. J. Crit. Care 2017, 38, 289–294.
- Genga, K.R.; Trinder, M.; Kong, H.J.J.; Li, X.; Leung, A.K.K.; Shimada, T.; Walley, K.R.; Russell, J.A.; Francis, G.A.; Brunham, L.R.; et al. CETP genetic variant rs1800777 (allele A) is associated with abnormally low HDL-C levels and increased risk of AKI during sepsis. Sci. Rep. 2018, 8.
- Levels, J.H.M.; Abraham, P.R.; Van den Ende, A.; Van Deventer, S.J.H. Distribution and kinetics of lipoprotein-bound endotoxin. Infect. Immun. 2001, 69, 2821–2828.
- Flegel, W.A.; Wolpl, A.; Mannel, D.N.; Northoff, H. Inhibition of endotoxin-induced activation of human monocytes by human lipoproteins. Infect. Immun. 1989, 57, 2237–2245.
- Baumberger, C.; Ulevitch, R.J.; Dayer, J.M. Modulation of endotoxic activity of lipopolysaccharide by high-density lipoprotein. Pathobiology 1991, 59, 378–383.
- Anderberg, S.B.; Luther, T.; Frithiof, R. Physiological aspects of Toll-like receptor 4 activation in sepsis-induced acute kidney injury. Acta Physiol. 2017, 219, 573–588.
- Habib, R. Multifaceted roles of Toll-like receptors in acute kidney injury. Heliyon 2021, 7, e06441.
- Zarbock, A.; Gomez, H.; Kellum, J.A. Sepsis-induced acute kidney injury revisited: Pathophysiology, prevention and future therapies. Curr. Opin. Crit. Care 2014, 20, 588–595.
- Lowry, J.L.; Brovkovych, V.; Zhang, Y.; Skidgel, R.A. Endothelial nitric-oxide synthase activation generates an inducible nitric-oxide synthase-like output of nitric oxide in inflamed endothelium. J. Biol. Chem. 2013, 288, 4174–4193.
- Tran-Dinh, A.; Diallo, D.; Delbosc, S.; Varela-Perez, L.M.; Dang, Q.B.; Lapergue, B.; Burillo, E.; Michel, J.B.; Levoye, A.; Martin-Ventura, J.L.; et al. HDL and endothelial protection. Br. J. Pharmacol. 2013, 169, 493–511.
- Graversen, J.H.; Castro, G.; Kandoussi, A.; Nielsen, H.; Christensen, E.I.; Norden, A.; Moestrup, S.K. A Pivotal Role of the Human Kidney in Catabolism of HDL Protein Components Apolipoprotein A-I and A-IV but not of A-II. Lipids 2008, 43, 467–470.
- Kronenberg, F.; Kuen, E.; Ritz, E.; König, P.; Kraatz, G.; Lhotta, K.; Mann, J.F.E.; Müller, G.A.; Neyer, U.; Riegel, W.; et al. Apolipoprotein A-IV serum concentrations are elevated in patients with mild and moderate renal failure. J. Am. Soc. Nephrol. 2002, 13, 461–469.
- Calabresi, L.; Simonelli, S.; Conca, P.; Busnach, G.; Cabibbe, M.; Gesualdo, L.; Gigante, M.; Penco, S.; Veglia, F.; Franceschini, G. Acquired lecithin: CHolesterol acyltransferase deficiency as a major factor in lowering plasma HDL levels in chronic kidney disease. J. Intern. Med. 2015, 277, 552–561.
- Aseem, O.; Smith, B.T.; Cooley, M.A.; Wilkerson, B.A.; Argraves, K.M.; Remaley, A.T.; Argraves, W.S. Cubilin maintains blood levels of HDL and albumin. J. Am. Soc. Nephrol. 2014, 25, 1028–1036.
- Zhong, J.; Yang, H.; Kon, V. Kidney as modulator and target of “good/bad” HDL. Pediatric Nephrol. 2019, 34, 1683–1695.
- Vaziri, N.D. HDL abnormalities in nephrotic syndrome and chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 37–47.
- Coughlin, M.M.; Babcook, J.; Prabhakar, B.S. Human monoclonal antibodies to SARS-coronavirus inhibit infection by different mechanisms. Virology 2009, 394, 39–46.
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369, 650–655.
- Wei, C.; Wan, L.; Yan, Q.; Wang, X.; Zhang, J.; Yang, X.; Zhang, Y.; Fan, C.; Li, D.; Deng, Y.; et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2020, 2, 1391–1400.
- Strollo, R.; Pozzilli, P. DPP4 inhibition: Preventing SARS-CoV-2 infection and/or progression of COVID-19? Diabetes Metab. Res. Rev. 2020, 36, e3330.
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, 1166–1171.
- Shen, W.J.; Azhar, S.; Kraemer, F.B. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu. Rev. Physiol. 2018, 80, 95–116.
- Kolleck, I.; Sinha, P.; Rüstow, B. Vitamin E as an antioxidant of the lung: Mechanisms of vitamin E delivery to alveolar type II cells. Am. J. Respir. Crit. Care Med. 2002, 166, S62–S66.
- Kolleck, I.; Schlame, M.; Fechner, H.; Looman, A.C.; Wissel, H.; Rüstow, B. HDL is the major source of vitamin E for type II pneumocytes. Free Radic. Biol. Med. 1999, 27, 882–890.
- Kreutz, R.; Algharably, E.A.E.H.; Azizi, M.; Dobrowolski, P.; Guzik, T.; Januszewicz, A.; Persu, A.; Prejbisz, A.; Riemer, T.G.; Wang, J.G.; et al. Hypertension, the renin-angiotensin system, and the risk of lower respiratory tract infections and lung injury: Implications for covid-19. Cardiovasc. Res. 2020, 116, 1688–1699.
- Lajoie, P.; Nabi, I.R. Regulation of raft-dependent endocytosis. J. Cell. Mol. Med. 2007, 11, 644–653.
- Baglivo, M.; Baronio, M.; Natalini, G.; Beccari, T.; Chiurazzi, P.; Fulcheri, E.; Petralia, P.; Michelini, S.; Fiorentini, G.; Miggiano, G.A.; et al. Natural small molecules as inhibitors of coronavirus lipid-dependent attachment to host cells: A possible strategy for reducing SARS-COV-2 infectivity? Acta Biomed. 2020, 91, 161–164.
- Li, G.M.; Li, Y.G.; Yamate, M.; Li, S.M.; Ikuta, K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect. 2007, 9, 96–102.
- You, C.H.; Hong, Y.S.; Kwak, J.Y.; Kim, N.Y.; Mee, S.R.; Jung, K.Y.; Lee, Y.H.; Kim, J.M.; Kim, J.Y. The relationship between ACE I/D polymorphism and HDL cholesterol. J. Prev. Med. Public Health 2006, 39, 505–510.
- Dou, X.; Li, Y.; Han, J.; Zarlenga, D.S.; Zhu, W.; Ren, X.; Dong, N.; Li, X.; Li, G. Cholesterol of lipid rafts is a key determinant for entry and post-entry control of porcine rotavirus infection. BMC Vet. Res. 2018, 14, 45.
- Korade, Z.; Kenworthy, A.K. Lipid rafts, cholesterol, and the brain. Neuropharmacology 2008, 55, 1265–1273.
- Radenkovic, D.; Chawla, S.; Pirro, M.; Sahebkar, A.; Banach, M. Cholesterol in Relation to COVID-19: Should We Care about It? J. Clin. Med. 2020, 9, 1909.
- Hu, X.; Chen, D.; Wu, L.; He, G.; Ye, W. Low Serum Cholesterol Level Among Patients with COVID-19 Infection in Wenzhou, China. SSRN Electron. J. 2020.
- Meher, G.; Bhattacharjya, S.; Chakraborty, H. Membrane Cholesterol Modulates Oligomeric Status and Peptide-Membrane Interaction of Severe Acute Respiratory Syndrome Coronavirus Fusion Peptide. J. Phys. Chem. B 2019, 123, 10654–10662.
- Garrido, P.F.; Calvelo, M.; Blanco-González, A.; Veleiro, U.; Suárez, F.; Conde, D.; Cabezón, A.; Piñeiro, Á.; Garcia-Fandino, R. The Lord of the NanoRings: Cyclodextrins and the battle against SARS-CoV-2. Int. J. Pharm. 2020, 588, 119689.
- Henrich, S.E.; McMahon, K.M.; Palacio, N.; Bhalla, P.; MacMaster, P.P.; Thaxton, C.S. Targeting scavenger receptor type B-1 (SR-B1) and cholesterol inhibits entry of SARSCoV-2 pseudovirus in cell culture. BioRxiv 2020.
- Glende, J.; Schwegmann-Wessels, C.; Al-Falah, M.; Pfefferle, S.; Qu, X.; Deng, H.; Drosten, C.; Naim, H.Y.; Herrler, G. Importance of cholesterol-rich membrane microdomains in the interaction of the S protein of SARS-coronavirus with the cellular receptor angiotensin-converting enzyme 2. Virology 2008, 381, 215–221.
- Cervin, M.; Anderson, R. Modulation of coronavirus-mediated cell fusion by homeostatic control of cholesterol and fatty acid metabolism. J. Med. Virol. 1991, 35, 142–149.


