1000/1000
Hot
Most Recent
 +1 point
+1 point
Myostatin, also known as growth and differentiation factor 8 (GDF-8), was identified in 1997 by McPherron and Lee.
Muscular dystrophies consist of a broad array of inherited conditions characterized by muscular wasting and atrophy. As clinical presentations in patients may vary due to a wide spectrum of phenotype–genotype variants for a particular gene, a common treatment, not depending on correcting a single molecular defect, has emerged as an attractive target for development. For the last 20 years, one of the most promising therapeutic subjects in the field of muscular dystrophies has been myostatin. Identified for the first time in 1997, myostatin knock-out in mice caused increased muscle mass [1] and mutations in the myostatin gene (MSTN) gene have subsequently been identified in the double muscled Belgian Blue and Piedmontese cattle [2][3][4] as well as whippet racing dogs [5]. In 2004, a loss-of-function mutation of MSTN in a German boy with a hypermuscular phenotype demonstrated that the effect of myostatin is functionally conserved across different mammalian species [6]. Since myostatin loss of function did not appear to have any negative impact on viability and longevity [7][8], interest was raised towards a novel treatment by harnessing the potential of inhibiting this negative regulator of muscular growth. Numerous studies in animal models and clinical trials have tried to explore this relationship with promising results in preclinical studies, which have translated poorly in human clinical studies.
Myostatin, also known as growth and differentiation factor 8 (GDF-8), was identified in 1997 by McPherron and Lee [1]. During embryogenesis, myostatin is expressed in the developing epaxial and hypaxial myotomes [9][10] and hereafter in muscular tissue postnatally, but has also been found at low expression in adipose tissue, heart and circulation throughout development [11][12]. As the mstn-gene is highly conserved among different vertebrate species [3][6][13][14][15][16], it is evident that it has an important function in muscle development and physiology, which has been preserved during the course of evolution. As a member of the TGF-β-superfamily, myostatin shows homology to other growth and differentiation factors, such as bone morphogenic proteins (BMP) and activins, which also elicit their biological function as dimers. Prepro-myostatin is synthesized as an N-terminal signal peptide followed by a propeptide domain and eventually a mature C-terminal domain [13]. During proteolytic processing, the signaling peptide is removed and the propeptide is cleaved from the mature protein. As the mature C-terminal domain dimerizes and forms disulfide bridges, it remains inactivated since noncovalent bonds between the mature dimer and the propeptide hold the mature myostatin in an inactive state [11][17][18][19]. To exert its function, the propeptide must be cleaved from the inactive complex by a family of BMP1/TLD-metalloprotease proteins [19][20]. Other than the propeptide itself, regulation of myostatin activity is also known to be mediated by follistatin [17][21], follistatin-related gene (FLRG) [11], Gasp-1 [22] and the proteoglycan protein decorin [23][24] typically by blocking the binding of myostatin to the receptors (Figure 1).
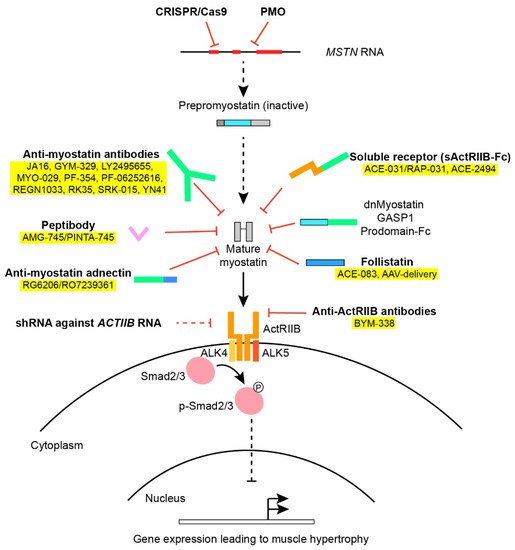
Figure 1. An overview of various approaches used in myostatin inhibition. Various factors and approaches in myostatin inhibition as outlined in Section 2 and Section 5. Treatments applied in clinical trials have been colored yellow. The Smad2/3 intracellular signaling pathway downstream the ActRIIB leads to altered gene transcription of muscle regulatory factors.
Once liberated from inhibitory proteins, myostatin, as well as other members of the TGF-β-family, binds to activin-receptors and, in the case of myostatin, mainly to the activin-receptor type IIB (ActRIIB) as well as the type IA receptor [17]. The activin receptors are transmembrane serine/threonine kinases that subsequently recruit and activate dimers of type I-receptors (ALK4 and ALK5) [25][26]. Depending on the receptor ligand and the composition of the receptor complex, the type I-receptor will phosphorylate and activate intracellular protein Smad2 and 3 downstream to the membrane receptors through the canonical Smad-pathway. Smad2/3 binds to Smad4 and the complex translocates to the nucleus [27], where muscle regulatory factors MyoD, Myf5 and Myogenin are repressed [28], preventing myoblast proliferation [29] and differentiation [28]. Obstruction of the myostatin pathway inhibits activation of Smad2/3, making Smad4 available in the BMP signaling pathway which promotes hypertrophy and counteracts the effects produced by myostatin [30]. Other noncanonical pathways activated by myostatin involve (among others) AMP-activated protein kinase (AMPK) and p38 mitogen-activated protein kinase (MAPK) [31][32].
Compared to healthy young men, there was no reported change in serum myostatin levels in an elderly population with mild or severe sarcopenia (as defined by muscular contractile force) [33]. Burch et al. reported that myostatin was 57% higher in a healthy cohort >25 years of age compared to a healthy cohort <25 years, with an age-dependent increase in the younger cohort but not in the older cohort [34]. A different study with more than 1100 participating men aged 20–87 years demonstrated that circulating myostatin level was dependent on age and body mass index [35]. Additionally, men had higher levels than women [34]. This is in contrast to findings of myostatin levels declining on ageing in men both measured by ELISA [36] and immunoplexed liquid chromatography with tandem mass spectrometry [37]. A smaller study of eight young and six elderly women showed higher levels of myostatin mRNA in muscle biopsies of the older group [38]. Various groups have sought to determine the use of myostatin as a potential biomarker for muscle wasting but the conclusions have been ambiguous [39][40][41].
The effect of age on the expression of not only myostatin but also other promyogenic muscle regulatory factors (MRF) following exercise was examined by Raue et al. They found that at rest, there is a relative upregulation of both MRF and myostatin prior to exercise in elderly women compared to younger ones, but that the postexercise downregulation of myostatin is not hampered by age [38]. A study in healthy and sarcopenic elderly men demonstrated that resistance training or a combination of resistance and endurance training caused a decrease in myostatin [42][43].
The clinical relevance of myostatin in humans was described for the first time in HIV patients, who had increased levels of myostatin compared to healthy subjects. Furthermore, the levels were even higher in the patients who met the definition of AIDS-wasting syndrome [13]. The role of myostatin in muscular atrophy and muscle wasting was also determined in mice that developed cachexia in response to myostatin overexpression [44]. Cachexia manifests as a complex metabolic syndrome due to an underlying illness characterized by muscle wasting in conditions such as chronic heart failure (HF), cancer, chronic obstructive pulmonary disease (COPD) or chronic kidney disease (CKD) [45]. The use of myostatin inhibitors in such populations with progressive muscle wasting or atrophy secondary to an underlying condition is attractive, as the preservation of muscle strength for ambulation, personal care and everyday independence is key in reducing morbidity and improving quality of life.
In terms of cardiovascular disease, the upregulation of myostatin in the cardiomyocytes surrounding an ischemic infarction in sheep was shown in 1999 [12] and myostatin protein and mRNA in skeletal muscle and myocardium were increased in a rat-model of volume overload heart failure [46]. Lenk and colleagues also found that the protein expression of myostatin was increased in the skeletal muscle and myocardium of a murine LAD-ligation heart failure model, which corresponded to later findings in chronic heart failure patients who had elevated levels of myostatin mRNA and protein in muscle biopsies compared to healthy controls [47][48]. The relationship between myostatin levels in the circulatory system and patients suffering from chronic heart failure has been examined by various groups. Increased myostatin levels in HF-patients could be expected, since impaired cardiac output reduces oxygen supply to the vascular bed of muscle tissue and less muscle means less oxygen consumption. As various studies have detected elevated [48][49][50], equal [51] or lower [40][52] levels of the latent and inactivated myostatin complex in the circulatory system, methodological differences in the detection of myostatin (e.g., Western blotting of promyostatin versus immunoassays of full-length myostatin and Liquid chromatography–mass spectrometry (LC–MS)) may account for these fluctuating results [34]. Furthermore, myostatin levels in decompensated chronic HF patients dropped upon compensation therapy, suggesting dynamics and variability in myostatin levels, which are sensitive to therapeutic interventions [53].
Treating cancer-associated cachexia by means of myostatin inhibition has been another field of interest. As myostatin was elevated in the gastrocnemius muscle of mice inoculated with the Yoshida AH-130 hepatoma [54], targeting the myostatin pathway seemed promising in preventing cancer cachexia. C26-tumor-bearing mice were treated with a soluble receptor of the ActRIIB (sActRIIB), which improved survival and muscle mass without reducing tumor size [55] and by treating the Lewis lung cancer-model with myostatin antibodies, muscular atrophy and loss of muscle force were attenuated [56].
COPD has been another target of interest due to the muscle wasting, since 30–40% of all people with COPD undergo muscle wasting as a secondary complication to impaired pulmonary function [57]. The link between myostatin and chronic hypoxemia was established in rats exposed to chronic hypoxia, which induced myostatin expression in rat muscle [58], and the increased the expression of myostatin in the vastus lateralis and serum of COPD-patients compared to healthy controls has also been described [58][59]. Later, serum myostatin was found to be significantly elevated in COPD-patients compared to controls but skeletal muscle mass only correlated negatively with serum-myostatin in males [60].
In CKD, myostatin is elevated in the serum and skeletal muscle of the rat model of CKD, (Cy/+), with increased activation of atrogenic transcription factors in EDL adding insights to the pathophysiology behind muscle wasting in this condition [61].
The effect of exercise on the expression of myostatin has been demonstrated numerous times. In a clinical study where subjects had immobilized a limb for two weeks following exercise rehabilitation, the casting-induced atrophy did not affect myostatin mRNA in muscle biopsies. However, exercise led to downregulated myostatin expression by approximately 48% [62]. These findings indicate that myostatin works in vivo by inhibiting hypertrophy, rather than inducing atrophy. Similar findings in exercise studies have been observed up to 24 h after exercise [63][64] and on protein-level in prediabetic patients performing moderate aerobic exercise for six months [65]. Most interestingly, “the myostatin paradox” was introduced by Kim et al., who in their exercise study discovered a positive correlation between myostatin mRNA and muscle mass [63], whereas the relationship would most intuitively be the opposite if not taking inhibitory factors into consideration. The authors speculate that high levels of myostatin transcripts in muscle might prime the muscles for additional growth.
The potential for the pharmacological regulation of muscular growth had to be explored in animal models of muscular dystrophy, atrophy and muscular regeneration before ultimately turning towards clinical trials in human subjects. We present here an overview of the various ways in which myostatin has been targeted in animal models. As myostatin inhibition has been utilized to examine various physiological processes other than merely muscular regeneration (including cancer survival, bone- and energy metabolism), the following will focus on the bulk of scientific work that describes the effect of myostatin on muscular tissue. Table 1 presents information, if available, on the animal model and genus, the pharmacological compound, muscle morphology, fiber-type specific changes, absolute and specific force amongst glycolytic and oxidative muscles, muscular stress resistance and histopathological improvements. This review is focused in particular on treatment-mediated functional improvements of muscle function, as these are essential for any translation to human clinical trials. Histopathological recovery, muscular growth and the upregulation of desirable growth factors or genes in vitro may be of less importance, as primary outcomes are invariably functional in preclinical studies and the degree of functional improvement ultimately decides whether a treatment will advance in additional preclinical or clinical investigations. Furthermore, increasing the absolute force is of interest to patients and clinicians who are looking for improvements in the activities of daily living, while the scientist will be looking for specific force (force per cross sectional area of a muscle) as an indicator of whether the underlying deficit has been compensated for.
Table 1. Results of previously published data from various means of myostatin inhibition in animal models.
| Species/Model | Compound | Muscle Morphology | Fiber-Type Specific Changes | Absolute Force/ Glycolytic |
Specific Force/Glycolytic | Absolute Force/ Oxidative |
Specific Force/Oxidative | Stress-Induced Force Drop | Histopathological Effect of Myostatin Inhibition | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Antibodies Blocking Myostatin | ||||||||||
| Mouse/wild-type (BALB/c, C57BL/6) |
JA16, ATA-842, mRK35, YN41, muSRK-015P, GYM-mFc | Fiber CSA increased in EDL [66] and Gas Increased weight of Gas, TA, Quad and TB, plantaris, Sol [67] |
Increased IIB fiber CSA, no effect on overall composition [68] | Increased grip strength [67][69][70] | [66][67][68][69][70][71] | |||||
| Mouse/mdx, Mouse/Sgcd−/−, Sgcg−/− |
JA16 | EDL: Increased weight and single fiber area [72][73]. Increase in TA, Quad, Gas [74] |
EDL: increased force | EDL: No effect | No effect | Sgcd−/−: No improvement in histopathology of TA, EDL, Gas and diaphragm albeit hydroxyproline reduced in TA. Fibrosis in diaphragm increased (Sgcd−/− [74]) and decreased (mdx [72]) |
[72][73][74] | |||
| Mouse/mdx | PF-354 | Increase in hindlimb muscle weight of 5 weeks treatment, no effect after 8 weeks. No effect/reduction in CSA |
Diaphragm: No effect | Diaphragm increased (young)/no effect (old) | No effect | Diaphragm: Increased fiber size in young animals, decreased fiber size in old animals | [75] | |||
| Mouse/mdx, TgCTA1D286G, Sod1G93A, A17, Rat/Sod1G93A Mouse/SmnΔ7 |
mRK35/RK35 muSRK-015P (in SmnΔ7) |
TA, Gas, Quad, EDL, diaphragm weight increased. Increased CSA in TA, EDL. No effect on weight or CSA in soleus [76] |
Quad: Increased proportion of IIB fibers [77] Increase in IIB fiber CSA, no effect in remaining fiber-types [78] |
TA, EDL: Increased force Plantarflexor group increased torque [78] |
TA, EDL: No effect effect on plantarflexor group [78] |
Gas: reduced atrophy, preserved fiber diameter. Diaphragm integrity preserved [79]. Reduced collagen I, III, IV deposits. No effect on intranuclear inclusion bodies [76][80]. Increased number of tubular aggregates [77]. |
[71][76][77][78][79][80] | |||
| Mouse/ CB17-SCID, C57BL/6, (Dexamethasone atrophy) |
REGN1033 | Increased weight in Gas and TA. Fiber area increased in Gas | No effect on fiber type composition | TA: Increased force | TA: No effect | [81] | ||||
| Monkey/cynomolgus | MYO-029, Domagrozu-mab, GYM-cyfc |
Increased muscular circumference | [70][71][82] | |||||||
| Myostatin Propeptide Administration or Overexpression | ||||||||||
| Mouse/mdx | Recombinant propeptide-Fc | EDL: weight, CSA, single fiber area increased | EDL: Increased force | EDL: Increased force | No effect | Decreased pathological changes | [83] | |||
| Mouse/mdx | AAV8- MPRO76AFc |
TA, Quad, Gas, Diaphragm increased | TA: Increased force | TA: No effect | Larger fibers, less fibrosis | [84] | ||||
| Mouse/calpatin 3-null mice (LGMD2A), Sgca−/− (LGMD2D) | rAAV2/1mSeAP-propmyoD76A | Increased muscle mass in calpain-3-null mice, no effect in Sgca−/−-mice | EDL: increased force (calpain-3-null mice) | EDL: No effect (calpain-3-null mice) |
Soleus: Increased force (calpain-3-null mice) |
Soleus: No effect (calpain-3-null mice) |
[85] | |||
| Soluble Receptor (sActRIIB-Fc) | ||||||||||
| Wild-type, C57BL/6 C57BL/10 |
ACE-031, sActRIIB, RAP-031 ACE-2494 |
Increased muscle weight. Fiber CSA increased in EDL [86] and in whole TA [87] |
Soleus: Type I and II-fiber CSA increased [88]. Quad; increased size of I, IIA, IIB-fibers. No fiber-type switch [89] |
EDL: twitch force increased, no effect on tetanic force [86]. Gas: no effect on max tetanic force [90] |
EDL: no effect [91][92] Gas: decreased [90] |
Soleus: increased force [91] | Soleus: no effect force [91] | [86][87][88][89][90][91][92][93][94][95] | ||
| Mouse/mdx | RAP-031, sActRIIB-Fc |
Muscle weight increased Diaphragm and triceps myofiber increased [96]. EDL single fiber CSA increased [97]. |
No fiber-type conversion [91] | EDL: increased force [97][98] | EDL: increased force [97], No effect [98]. EDL, TA decreased force in older animals |
Soleus decreased force | Soleus decreased force | No effect | Diaphragm, TA: No effect on histopathology, hydroxyproline [93][97]. Fibrosis decreased [96]. No visible effects on H/E pathology. SDH stains without effect of treatment [99]. eMHC: no effect [93]. |
[91][93][96][97][98][99] |
| Mouse/TgActa1H40Y, Mtm1R69C, Mtm1δ4, R6/2, Dysf−/−, Cav3P104L |
RAP-031, sActRIIB-Fc |
Increased muscle weight, increased fiber size | Quad: oxidative fiber diameter increased. Diaphragm: glycolytic myofibers hypertrophy [100]. IIB fiber hypertrophy, no fiber type switch [89][101] |
No effect [100]. EDL, TA increased force [102] |
No effect [100] | No effect [100] | No effect [100] | Nemaline rod structures unchanged [100]. Gross evaluation of diaphragm: unaffected by genotype or treatment [89]. Fibrotic changes improved |
[89][100][101][102][103][104] | |
| Anti-ActRIIB Antibody | ||||||||||
| Mouse/SCID | BYM338 | Increased weight of TA, EDL, Gas. Soleus increased weight (in high dose) [105] |
Gas: increased force [106] | [105][106] | ||||||
| Mouse/C57BL/6 (glucocorticoid-induced atrophy) | BYM338 | TA weight and CSA increased | TA increased force | [105] | ||||||
| Follistatin Administration or Overexpression | ||||||||||
| Mouse/ F66;Dysf−/−, F66;mdx |
Follistatin overexpression | Muscle mass maintained in F66;mdx, decreased in F66;Dysf−/− | F66;Dysf−/−: EDL: Decreased force | F66;Dysf−/−: Exacerbation of dystrophic features. Increased Evans Blue Dye (EBD) uptake F66;mdx: Dystrophic features not exacerbated, mild improvement |
[103] | |||||
| Mouse/mdx, Sod1G93A | AAV-delivered follistatin i.m. | Increased weight of TA, Gas, Quad, triceps | Increased grip strength | Young mdx: increased myofiber size. Satellite cell markers: no diff Old mdx: Fever necrotic fibers and mononuclear infiltrates |
[107][108] | |||||
| Monkey/Cynomolgus | AAV-delivered follistatin i.m | Increased fiber size | Quad: Increased force | Myofiber hypertrophy | [109] | |||||
| Mouse/C57BL10, mdx, | ACE-083 | Increased CSA, weight | TA: increased force | TA: no effect | [110] | |||||
| Mouse/C57BL/6 | FS-EEE-mFc and FST288-Fc | Increased muscle weight | [98][111] | |||||||
| Mouse/mdx | FS-EEE-mFc | Increased weight in gas, Quad, triceps, TA | EDL: Increased force | EDL: No effect | Decreased necrosis and fibrosis in Quad, no effect in diaphragm | [98] | ||||
| Liver-mediated Overexpression of Dominant-negative Myostatin (dnMSTN), sActRIIB and Myostatin Propeptide | ||||||||||
| Mouse/MF-1 (wild-type) | AAV8 over-ekspression (propeptide) | Gas, TA increased mass. EDL and soleus increased CSA. | Increased CSA of type I, IIA and IIB-fibers | EDL: No effect | EDL: No effect | Soleus: increased force | Soleus: No effect | [112] | ||
| Mouse/SmaC/C | AAV-mediated systemic expression (dnMSTN and sActRIIB) | Increased weight in TA, Gas, Quad. dnMSTN-cohort: Increased CSA in EDL and TA but not in soleus |
TA: Increased IIA size EDL: Increased IIA and IIB size and total fiber number. Soleus: No effect vs. controls. I-fibers generally unaffected |
EDL increased vs. SMAC/C control | EDL; Decreased force | Soleus: Increased force | Soleus: No effect | [113] | ||
| Mouse/mdx | AAV-delivered liver-specific promoter: dnMSTN, sActRIIB | Increased weight in TA, Gas, Quad, EDL, Soleus EDL: increased CSA Soleus: No effect in weight [114] |
EDL: IA + IIB increased fiber size. Increased proportion of IIB fibers in EDL and Soleus. Soleus: Increased size and proportion of IIA-fibers Diaphragm: IIX fibers proportion increased, IIA fibers proportion decrease [115] Diaphragm: No effect in specific fiber-type size [114] |
EDL: increased force | No effect (decreased force by 10 months of treatment) | Soleus: increased force | Soleus increased force [115] Soleus no difference [114]. Diaphragm: no effect |
[114][115][116] | ||
| Dog/GRMD | AAV-delivered liver-specific promoter (dnMSTN) | Increased weight in Tibialis cranialis, EDL, Gas, flexor digitorum superficialis | Increased size of IIA-fibers, no effect in I-fibers. No fiber type switch |
[117] | ||||||
| RNA Interference and Anti-oligonucleotides against Myostatin or ActRIIB | ||||||||||
| Mouse/mdx | Antimyostatin PMO | No effect in weight of diaphragm, EDL, Gas, Soleus, TA | Diaphragm: no difference in fiber-type content (I, IIA, IIX, IIB) | Diaphragm and TA: no effect on fiber diameter and collagen IV content | [118] | |||||
| Mouse/mdx (female) | AAV-delivered shRNA, i.m. | TA: No effect on CSA, fiber number increased | TA: No effect | TA: No effect | [119] | |||||
| AAV-Cas9-mediated Myostatin Gene Editing | ||||||||||
| Mouse/C57/BL10 | rAAV-SaCas9 | Increased fiber area and number of fibers per area | [78] | |||||||
| Myostatin Knock-out/Crossbreeding | ||||||||||
| Mouse/Mstn−/− | Increased muscle weight vs. wild-type. Increased fiber number and CSA of EDL and soleus [120] |
EDL fiber-type composition: IIA and IIX incidence decreased, IIB increased in EDL and TA. Soleus CSA increased only in IIA-fibers [121] |
EDL: Increased [120]/no effect [121][122] |
EDL: Decreased |
Soleus: Increased | Soleus: No effect | EDL: Force deficit Soleus: No force deficit |
Decreased hydroxyproline content in EDL, no effect in soleus [120]. Cytoplasmic inclusions of tubular aggregates in older mice [122] |
[120][121][122][123][124][125][126] | |
| Mouse/BehC/C | Increased muscle weight | EDL: No effect [122] | EDL: Decreased force [122] | [122][127] | ||||||
| Mouse/ Mstn−/−;mdx Mstn−/−;Sgcd −/− MstnPro;Cav3P104L |
Increased mean fiber diameter and muscle weight [104][128][129] | Mstn−/−;mdx: Reduced fibrosis [128] Mstn −/−;Sgcd −/−: Hydroxyproline content decreased in EDL [74] |
[74][104][128][129] | |||||||
| Mouse/Mstn−/−; dyW/dyW | Increased muscle mass, muscle CSA and fiber CSA. (increased mortality) |
Decreased type I fiber composition | No effect on necrosis, inflammation or infiltrating cells. Less fat tissue. | [130] | ||||||
Mainstream results from various antimyostatin treatments in animal models. Specific results that were distinct for a particular study and not general for all of the references have been titled as such. Abbreviations: AAV; adeno-associated virus, ActRIIB; activin receptor type IIB, CSA; cross-sectional area, EDL; m. extensor digitorum longus, eMHC; embryonic myosin heavy chain, Gas; m. gastrocnemius, GRMD; golden retriever muscular dystrophy i.m.; intra-muscular injection, LGMD; limb-girdle muscular dystrophy, Quad; m. quadriceps, SDH; succinate dehydrogenase, TA; m. tibialis anterior, TB; m. triceps brachii.
The pharmacological approaches to inhibiting myostatin activity in vivo have included: (a) systemic administration of antibodies against myostatin; (b) overexpression or administration of the myostatin propeptide; (c) systemic administration of the activin-IIB-receptor itself; (d) administration of antibodies directed against ActRIIB; (e) overexpression or administration of follistatin; (f) liver-mediated overexpression of a soluble receptor (sActRIIB), dominant-negative myostatin (dnMSTN) or the propeptide; (g) RNA interference and antioligonucleotides against myostatin or ActRIIB or; (h) AAV-Cas9 mediated myostatin gene editing. Finally, we have also included works on the effects of transgenic knock-out models and crossbreeding with the preexisting models of muscular dystrophy (i).
Bogdanovich et al. were the first to successfully treat the commonly used mouse model of Duchenne Muscular Dystrophy (DMD), the mdx, with antibodies directed towards myostatin (monoclonal antibodies, JA16) [72]. The results were promising, as the diaphragm and the skeletal muscle, which in the mdx reproduces the pathological features seen in muscles of DMD-patients most accurately [131], showed fewer degenerative features compared to controls. Meanwhile, m. extensor digitorum longus (EDL) had increased weight, cross-sectional area (CSA) and absolute force but failed to show improvement on specific force and stretch resistance. Similar results with increased muscle weight and absolute force but lack of improvement in specific force and resistance were seen in the Sgcg−/− model of limb-girdle muscular dystrophy (LGMD) 2C in a design of similar age and treatment length to the previously mentioned study [73]. The Sgcd−/− mouse model of LGMD2F also treated with JA16 antibodies was not able to improve fibrosis in either young or older Sgcd−/− animals (4 and 20 weeks old at treatment start, respectively) with older animals even showing signs of worsening of fibrosis [74]. Interestingly, a 5-week treatment period of very young (16 days old) mdx-animals showed positive effects on the diaphragm, as specific force increased while absolute force was unaffected, fiber size increased and connective tissue infiltration of the diaphragm was reduced [75], indicating that early initiation of treatment is crucial for a positive effect. Another monoclonal antibody developed by Pfizer, mRK-35, was also able to increase absolute but not specific force in mdx mice [71] and the TgActa1D286G mouse model of nemaline myopathy [77]. Treatment of the Sod1G93A mouse and rat models of amyotrophic lateral sclerosis (ALS) with RK35 improved grip strength compared to placebo controls but did not delay disease onset or extend survival of either model [79]. Later, Muramatsu and colleagues introduced the concept of “sweeping antibody technology” with the GYM329-antibody designed to bind and clear latent myostatin from the circulatory system, which increased muscle mass in three mice models and cynomolgus monkeys and also improved grip strength in the mice [70]. As opposed to other antimyostatin antibodies, GYM329 did not bind GDF-11 and this specificity appears to induce an enhanced effect on muscle mass in treated animals. Especially in older animals, where other myostatin inhibition treatments fail or struggle to achieve an effect, GYM329 appeared superior. Other models of neuromuscular disorders such as the SmnΔ7 mouse of spinal muscle atrophy (SMA) had increased absolute muscle torque but not specific torque after treatment with the muSRK-015P antibody versus myostatin (combined with salvation of Smn2-gene mRNA) [132]. In a study of micro-gravity-induced muscular atrophy, mice were held at the International Space Station and treated with YN41 for 6 weeks, inducing improved grip strength compared to controls, as well as increased muscle mass [67].
As previously mentioned, the myostatin propeptide functions as an inhibitor of myostatin, as it binds myostatin in an inactive complex. Propeptide-based inhibition by intraperitoneal injection for three months resulted in increased body mass, EDL mass, absolute and specific force in EDL. There was no effect on stretch-resistance but the histopathological phenotype of the diaphragm improved compared to untreated mdx [83]. Bartoli et al. treated calpain-3-null and Sgca−/−-mouse models of LGMD2A and 2D, respectively, by local and systemic overexpression of the propeptide but were only able to improve the calpain-3-null mice [85].
In order to increase the specific targeting of myostatin and reduce binding of the variety of other ligands that also bind to and activate ActRIIB, another approach based on the systemic administration of a soluble activin type IIB receptor, sActRIIB, has been widely utilized. The compound RAP-031, developed by Acceleron, is a fusion protein consisting of the extracellular domain of the ActRIIB linked to the Fc-portion of murine IgG to delay systemic clearance. Applying this approach, Pistilli and colleagues demonstrated an increase in both absolute and specific force of EDL in mdx mice [97], a functional outcome, which unfortunately has been difficult to replicate in both wild-type [86][92], mdx [91][93][98], and nemaline myopathy mouse models [100]. The treatment of mice with muscular atrophy due to spinal cord injury with RAP-031 did not alleviate the atrophy [133]. A hypoxia model in wild-type mice showed improved resistance to eccentric lengthening but no other studies using the soluble receptor have shown improvements to stretch resistance [134]. The specific hypertrophy of fibers with a IIB fiber-type composition was observed in two models of myotubularin-deficient mice [89][101] but also in other fibers of wild-type animals [88][89].
Blocking the ActRIIB itself by antibodies has not been widely used as another means of myostatin inhibition. Novartis developed BYM338 (bimagrumab, which would progress into clinical trials as mentioned below) and described the receptor-specificity in cell cultures and myoblasts while also showing the effects on body and muscle mass in both SCID-mice and a glucocorticoid atrophy model [105].
Like the myostatin propeptide, follistatin is able to inhibit not only myostatin but also shows affinity for other TGF-β-family members (such as BMPs and activins) [22][135]. Transgenic overexpression of follistatin primarily showed increased muscle weight and fiber diameter [17]. Transgenic mice overexpressing a follistatin-derived myostatin inhibitor crossed with the mdx ameliorated the dystrophic features in terms of grip strength and pathohistological features [136]. When transgenic overexpression of follistatin (F66-mice) is crossed with the dysferlinopathy LGMD2B model Dysf−/−, the positive effect on muscle weight in F66;Dysf−/−-mice declines with age and the specific force of EDL is reduced, compared to F66-mice, exacerbating the dystrophic phenotype [103]. Furthermore, ActRIIB-FC-administration in Dysf−/−-mice ameliorated histopathological changes, but increased creatine kinase (CK, a marker of muscular damage and membrane integrity) levels. The authors conclude that follistatin overexpression accelerated the degenerative features in the dysferlinopathy model, as the dystrophin-deficient mdx was not exacerbated [103], and suggest that muscle hypertrophy may have pernicious effects depending on the disease context.
Another approach using a follistatin-based fusion protein ACE-083 by local intramuscular injections increased CSA, weight and absolute, but not specific, force of injected muscle tibialis anterior (TA) in the Trembler-J mouse model of Charcot–Marie–Tooth disease and mdx [110].
As the systemic clearance of follistatin is rather quick, systemic versus local administration poses a challenge. Thus, the pharmacokinetic properties were edited and a long-acting follistatin-based molecule (FS-EEE-hFc) was engineered by Shen and colleagues [137] and applied by intravenous and subcutaneous administration to wild-type and mdx-animals [98]. The subcutaneous treatment of young (4 weeks) mdx-mice for 12 weeks also undergoing an exhaustion-exercise regime showed increased muscle weight and absolute but not specific force increments [98].
Using the same approach as mentioned earlier with adeno-associated virus 8 (AAV8)-delivered myostatin inhibitors, Morine and colleagues treated the mdx with AAV-vectors, which brought liver-mediated transcripts of sActRIIB [114] or dnMSTN [115] into circulation. The sActRIIB treatment did increase the muscle mass, fiber size and absolute force of the EDL, while CK decreased. However, there were no positive effects in soleus or specific force [114]. The dnMSTN paper showed that the treatment in mdx-mice was predominantly observed in the fast fibers (IIA, IIX and IIB) of both the EDL and soleus, while soleus increased both absolute and specific force and CK decreased [115]. A similar study in the D2.mdx only reported beneficial effects on absolute force in EDL [116].
Similar to the treatment regimes in the mdx, Liu and colleagues treated the C/C mouse model of spinal muscle atrophy (SMA) with AAV8-vectors containing transcripts for dnMSTN and sActRIIB, respectively [113]. Both treatments increased the size of type IIA and IIB-fibers, leaving type I-fibers unaffected (IIX was not measured). While specific force was unaffected by treatment, absolute force increased in EDL (both treatments) and soleus (only sActRIIB).
Another approach was used in wild-type MF-1 mice, where propeptide coupled to an immunoglobulin Fc molecule was delivered by means of AAV8 vectors to hepatocytes, ensuring an intrinsic production of the inhibitor [112]. In contrast to exogenic injections of the propeptide, Foster and colleagues treated mice from six weeks of age and found an increased absolute force in oxidative muscle soleus but not in EDL. Both EDL and soleus increased CSA, as well as subanalyses of fiber-types I, IIA and IIB. In a similar design, mdx-mice were treated at the age of three months, which increased body mass, grip strength, muscle mass and fiber radius [84]. The absolute twitch and tetanic force production improved but specific force did not.
Myostatin has also been sought downregulated by means of RNA interference. Dumonceaux et al. combined short hairpin RNA (shRNA) interference of ActRIIB mRNA with AAV mediated exon-skipping of dystrophin. The number of fibers increased in TA, but force production was unchanged in mice that received myostatin interference solely compared to untreated mdx [119].
In contrast to AAV-mediated gene therapy, antisense oligomers (AOs) hold no risk of uncontrolled genome insertion and levels of exon skipping can be regulated or aborted over time. Antisense phosphorodiamidate morpholine oligomers (PMOs) causing exon-skipping of myostatin increased TA weight and CSA locally in mdx-mice [138]. A follow up study combining systemic treatment with two different PMOs that restored dystrophin and inhibited myostatin, respectively, was promising but the mdx mice receiving the myostatin-inhibiting PMO did not benefit from this treatment alone [118]. A similar study demonstrated similar increases in muscle mass in PMO-skipped myostatin, but also demonstrated that skipping varied among muscles, with the highest level of skipping in the soleus. These studies emphasize the importance of the design of the PMO, as well as the variable results obtained in healthy and mdx animals, suggesting that histopathology plays a role in efficiency of the treatment [118][138].
Recently, it was demonstrated that myostatin knock-out by the means of AAV-Sa-Cas9 gene editing delivered by intramuscular injections increased fiber area and number of fibers per area in aged wild-type mice [78]. However, functional outcomes were not described.
The murine hypermuscular myostatin knockout (Mstn−/−, also denominated ‘the myostatin-null’) described in 1997, has subsequently been further examined and crossed with various mouse models of neuromuscular diseases. The myostatin-null itself has been described numerous times [1][122][126] with increased muscle and body mass. Force measurements have shown both positive and no effect on absolute force in the myostatin knock-outs but decreased specific force has generally been reported [120][121][122]. An increased proportion of fast fiber-types has been the common observation [122][123][124][125][126], in line with the findings in studies of pharmacological myostatin inhibition (see above). Another model of myostatin malfunction includes the Compact-mouse (also known as the Berlin High Line BEHC/C), which contains a 12-bp deletion in the propeptide domain of promyostatin (MstnCmpt-dl1Abc) but leaves the biologically active growth-factor domain of myostatin unaffected [139]. Kocsis and colleagues later found that the Compact genetic background itself, in addition to the promyostatin genetic deletion, determines the phenotype [127] and the use of this model has been rather limited.
A third mouse model is the lean myostatin mouse (Mstnln/ln), which has an induced loss-of-function mutation leading to a peptide without the ligand, thus a complete lack of myostatin. This model has similarly increased muscularity but has had most of its use in the field of metabolic research [140].
Crossing myostatin-null with other models of muscular dystrophy has occasionally been the preceding study to pharmacological interventions. Crossing myostatin-null with mdx [128][129] or caveolin-3-deficient mice with transgenic mice overexpressing the myostatin prodomain (“MSTNPro”) [104] did ameliorate the pathological features by increasing body weight, fiber numbers and improving grip strength. However, the crossing of myostatin-null mice with the dyW/dyW laminin-deficient mouse model of congenital muscular dystrophy failed to improve the dystrophic phenotype and postnatal lethality was even increased [130].
In addition, a recent study crossing a follistatin overexpressing mouse strain with the calpain 3 knock-out mouse model for LGMD2A led to increased glycolytic muscle mass, but caused the loss of AMP-activated protein kinase signaling, important for contraction-induced glycolysis and poor exercise tolerance [141].





