2. Bruton’s Tyrosine Kinase, Structure and Regulation
Protein kinases (PTKs) act like a person flipping a molecular switch to control whether proteins are turned on or off. Kinases accomplish this by facilitating the transfer of a phosphate group from ATP to either a serine, threonine or tyrosine residue of their target protein
[7][8]. BTK is one of the five Tec family kinases, a group of cytosolic kinases that phosphorylate tyrosine residues. The other four Tec family kinases include: Txk/Rlk, Itk/Emt/Tsk, Bmx and Tec, the enzyme which the family was named after
[9]. The Tec family of kinases are one branch of the Src module kinases that mediate an array of cellular signaling processes. The other Src module kinases include ABL, CSK, FRK and BRK families
[8].
Structurally, the SRC root module that the Tec family of kinases stem from consists of a single polypeptide chain comprising an SH3 and an SH2 domain. The SH2 domain is connected by an SH2-kinase linker to the kinase domain (AKA; SH1 domain), which is ultimately responsible for catalytic activity
[8]. In BTK’s inactive state, the SH2 and SH3 domains impinge upon the kinase domain, thereby stabilizing inactivity and providing allosteric control over the catalytic site. The hallmark feature of the Tec family kinases that differentiates them from their relative kinases is the presence of the amino terminal Pleckstrin Homology (PH) domain that also confers allosteric control by further stabilizing the inactive conformation
[9]. Following the amino PH domain is a proline-rich Tec homology (TH) domain, followed by the Src domains SH3, SH2 and the carbonyl SH1 domain
[7] ().
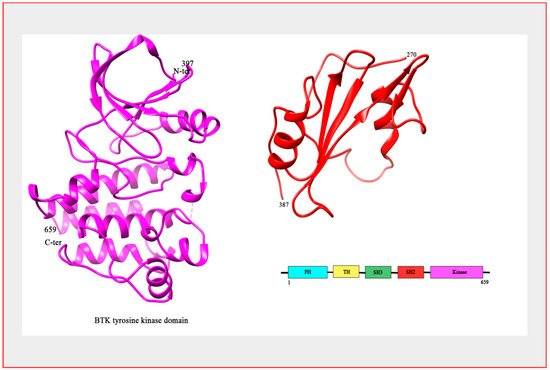
Figure 1. Structure of BTK. The crystal structure of BTK kinase domain (colored magenta; PDB: 3GEN) and SH2 domain (colored red; PDB: 2GE9). Bar Diagram representing the domain organization of the BTK protein.
Genetically, BTK has been shown in humans to be on the long q arm of the X chromosome at location Xq22.3-q22. The gene encompasses 37.5 kb, contains 19 exons and is expressed in all hematopoietic cells with the exception of T cells and possibly plasma cells
[10]. Loss of the X chromosome has been reported in MM; however, these cases are leading to monosomy in females, therefore complete loss of BTK coding does not occur. There is little data supporting amplification of the BTK coding portions of the X chromosome in MM.
BTK is primarily cytosolic but can be recruited to the cell membrane due to interactions with its PH domain. The PH domain confers the ability to bind to phosphoinositides, thereby merging the tyrosine kinase and the phospholipid signaling systems
[9][11]. BTK activation has been shown to be directly linked to its initial membrane recruitment and subsequent phosphorylation. Following its recruitment to the membrane, BTK activation occurs in two steps. Step one is the trans-phosphorylation of BTK on tyrosine residue at position 551 (Y551) in the kinase domain by either Lyn, SYK or other Src family kinases
[12]. This first step is thought to be responsible for the enzymes’ catalytic ability and consequently results in the second step, autophosphorylation of tyrosine at position 223 (Y223) in the SH3 domain
[13]. The Y223 position of the SH3 domain is highly conserved among signaling proteins and is thought to lie in the surface of the ligand-binding groove, which implies that this autophosphorylation may play a role in protein-protein interactions as well as stabilizing the active form, although the extent of these interactions is not fully understood
[7][13].
BTK activation can occur through other Src kinases but is primarily regulated by its transient recruitment to the cell membrane through the canonical PH-TH module with phosphatidylinositol (3,4,5)-trisphosphate (PIP
3) membrane lipids and subsequent phosphorylation. Little is known about the structural mechanism of this membrane interaction. However, recent simulations suggest that PIP
3 acts as a regulator of BTK activity as it controls PH-TH Saraste dimerization within the cell membrane, which may be an important step in enzymatic activation
[13][14][15]. BTK catalytic activity and tyrosine phosphorylation increases in response to cross linking or stimulation of the sIGM complex, the IL-5 or IL-6 receptors of B cells and the IgE receptor of mast cells, all of which are over expressed in MM patients. BTK activation then triggers activation of protein kinase B (AKT), phospholipase c (PLC) and calcium mobilization, leading to increased downstream effects including proliferation, differentiation and cell survival. Thus, chronic activation of BTK through the tyrosine kinase or phospholipid signaling systems can be a key driver in several neoplastic malignancies and a valuable therapeutic target in many more
[16].
3. BTK as a Potential Target in Malignancies
The phosphorylation of proteins is by far the most common form of reversible post-translational modifications that modulate the activity of biochemical signaling pathways in humans
[17]. The PTKs alter specific substrate activity and/or interactions with other proteins and are a fundamental method of controlling the cell cycle. PTK inhibition (PTKi) has proven to be an effective and accessible way to favorably alter the cellular signaling underlying hundreds of pathologies, including cancer
[7][17]. This is largely due to the abnormal activation and dysregulation of these kinase mediated pathways which act as driving forces in many malignancies. In cancer cells, this abnormal activation can lead to increases in proliferation, stemness, survival, migration, motility and metabolism, all of which are associated with less favorable outcomes in patients
[7].
The BTK pathway is universal in blood cells, as its signaling molecules are expressed in all hematopoietic lineages
[18]. BTK in particular plays a crucial role in the development of peripheral B-cells as well as the survival of leukemic cells and their interaction with the BMM
[7][19]. BTK is located in the cytoplasm of B-cells and is required for proper B-lymphocyte development, differentiation and signaling
[11]. When the gene encoding BTK is mutated, a condition called X chromosome-linked agammaglobulinemia (XLA) results. This condition is characterized by minimal B cell populations and ultimately results from altered B cell receptor (BCR) signaling and high rates of apoptosis. Clinically, patients with XLA exhibit impaired immune function, reductions in all classes of serum immunoglobulins, markedly reduced B cell lymphocytes populations, severe neutropenia, and an inability to make antibodies in response to vaccines and repetitive bacterial infections that onset several months after infancy
[20]. Due to this relation, the kinase itself is named after the Pediatrician Ogden Bruton, who reported the first case of XLA in 1952 ()
[21].
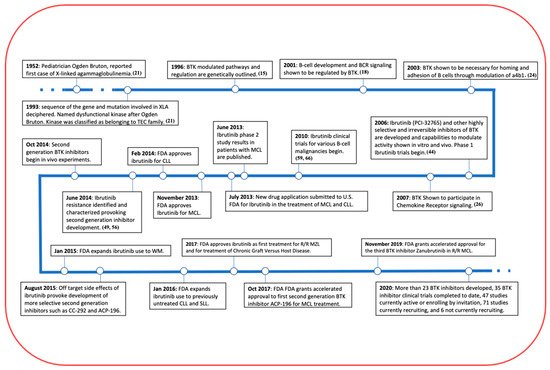
Figure 2. A Timeline of BTK and its inhibitor. BTK has made remarkable progress in the past 10 years in its treatment in a variety of B cell related malignancies and even more clinical trials are underway, including some in MM.
BTK plays an important role in various blood cell signaling pathways besides BCR signaling, including chemokine receptor 4 (CXCR4) signaling, toll-like receptor (TLR) signaling and Fc receptor signaling. Many of these signaling pathways result in common downstream effects due to the activation of BTK. For instance, BCR and CXCR4 activation of BTK have both been implicated to be important in controlling the adhesion of vascular cell adhesion proteins (VCAM)
[7][22]. A comprehensive outline of how BTK interacts in each of these pathways and the implications it has in neoplasm is provided below.
B Cell Receptor Signaling: Due to BTK’s causal role in the B cell disorder XLA, the most thoroughly understood pathway which it mediates is B Cell Receptor (BCR) signaling
[7]. In the germinal center, BCR, TLR and CD40 signaling are recognized as playing a fundamental role in the differentiation of B cells to plasma cells. After activation of B cells through BCR, TLR and/or CD40, upregulation of interferon regulatory factor 4 (IRF4) expression and downregulation of B-Cell Lymphoma 6 (BCL-6) expression occurs in addition to a loss of PR/Set Domain 1 (PRDM1) repression, resulting in down regulation of Paired Box 5 (PAX5) gene and upregulation of X-Box Binding Protein 1 (XBP1) and ultimately resulting in a loss of the B cell gene activation and phenotype. Furthermore, in the centrocyte region, stimulation of Interleukin 10, 21 or 6 (IL-10, IL-21 or IL-6) results in signal transducer and activator of transcription 3 (STAT3) activation, yielding PRDM1 overexpression and hence the gain of the plasma cell phenotype ()
[3].
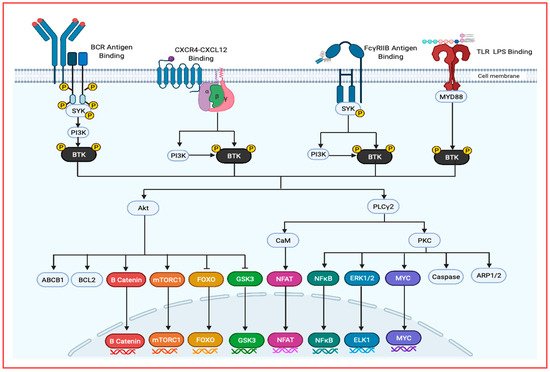
Figure 3. An overview of BTK activators and downstream signaling targets. BTK can be activated through several pathways include BCR signaling, chemokine signaling, Fc receptor signaling and TLR signaling. Following BTK integration to the cell membrane through its PH domain interaction with PIP3, the phosphorylated protein becomes cytosolic. These signaling pathways lead to the activation of AKT, PLCy2 and a protein complex including BTK, IRAK(1,2,4) and more leading to the downstream transcription factors that promote cancer growth, survival, stemness and migration.
BTK is essential in the B-cell receptor signaling of peripheral B cells, as it inhibits apoptotic signals and controls proliferation, differentiation, and adhesion. In the absence of BTK, peripheral B cells have a high rate of apoptosis, corresponding to BTK’s role in induction of anti-apoptotic protein Bcl-xL. Following BCR antigen engagement, either SYK or Lyn are required to initiate the formation of a signalosome at the cell membrane, allowing for efficient signal transduction. The signalosome also contains; Cbl-interacting protein of 85 kDa (CIN85), the SH2 containing leukocyte protein of 65 kDa (SLP65), and B cell linker (Blnk)
[23]. SLP65 acts as a scaffold protein orientating several signaling proteins including SYK, BTK and phospholipase Cγ2 (PLCγ2). Through this complex, SYK phosphorylates BTK, thereby allowing BTK to activate PLCγ2 and initiate a cascade by activating four separate families of different non-receptor protein kinases, resulting in key cell proliferation and survival signaling events. These activated kinases include PLCγ2, mitogen-activated protein kinase (MAPK), protein kinase B (AKT) and components of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway (). These kinases activate several notable downstream targets with direct impacts on increasing cell survival rate, drug resistance, stemness and ultimately relapse in the case of MM.
After phosphorylation of PLCγ2 by BTK, PLCγ2 can hydrolyze phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 modulates intracellular calcium levels and thereby can activate nuclear factor of activated T-cells (NFAT) transcription through calcineurin and calmodulin. DAG mediates activation of PKCβ, which can induce activation of several members of the MAPK family, including extracellularly regulated kinases 1 and 2 (ERK1/2), c-jun N-terminal kinase (JNK), p38 and NF-κB ().
On the other hand, AKT activation by BTK leads to downstream activation of proliferation and pro-survival protein pathways such as mammalian target of rapamycin (mTOR), NFAT, Forkhead Box O proteins (FOXOs) and NF-κB.
Besides the kinases it activates, the BCR has also been shown to play a role in controlling integrin α4β1 (VLA-4)-mediated adhesion of B cells to VCAM and fibronectin, alluding to the role BTK can play in the metastasis of B cell malignancies and their tumor-stroma interactions. BTK directly interacts with Wiskott-Aldrich syndrome protein (WASP), leading to its activation which induces RAC-dependent actin filament rearrangements. This has been shown to be dependent on BTK, PLCγ2 and PKCβ but not ERK1 or ERK2
[7][24].
Chemokine Receptor Signaling: Chemokines are secreted proteins that function as a leukocyte-specific chemoattractant. CXCR4 is one of the many chemokine receptors, and is a G coupled protein receptor expressed in many hemopoietic and non-hemopoietic cell types. CXCR4 has been studied intensively; it binds stromal cell-derived factor 12 (CXCL12), CD4 and CD74, it is overexpressed in 23 human cancers and plays important roles in immune response, hematopoiesis and pro-tumorigenicity.
When CXCR4 binds its endogenous ligand, CXCL12, which is highly expressed in the bone marrow, the trimeric G-protein subunits of CXCR4 dissociate and activate several pathways involved in chemotaxis, intracellular calcium flux, cell adhesion, survival, proliferation and gene activation. Therefore, CXCR4 activation in neoplasm is linked to increased proliferation, metastasis, angiogenesis, drug resistance and ultimately relapse. Accordingly, blocking the receptor has been shown to have therapeutic effects in MM including the disruption of tumor-stroma interactions, reduced proliferation, sensitization to cytotoxic agents, impaired homing and inhibited metastasis
[25][26]. Several studies have shown that in MM, CXCR4 is aberrantly activated and associated with the progression, spread and relapse of the cancer. CXCR4 activation in MM is shown to be induced by several factors including hypoxia, TNF-α, TNF-β and VEGF.
BTK is an important signal transductor in CXCR4-CXCL12 signaling as BTK inhibition has been shown to deregulate CXCR4 expression and signaling
[27]. After CXCL12 has bound to CXCR4, the Gα and Gβ subunits activate PI3K. PI3K activation then interacts with the PH domain of BTK, recruiting it to the cell membrane, allowing for BTK activation and leading to PLCγ2, AKT, MAPK and NF-κB pathway activation. In addition to PI3K activation of BTK, the Gα and Gβ subunits can both directly bind to BTK at the PH and TH domains and the Gα subunit can activate BTK directly. When BTK is inhibited, it has been shown to inhibit CXCL12 and CXCL13 induced phosphorylation of PLCγ2, ERK1, ERK2, JNK and AKT proteins, proving BTK’s role in this major pathway (see )
[7][28].
Toll-Like Receptor Signaling: Toll–like-receptors (TLRs) are membrane bound receptors that mediate the innate immune system and are activated by binding ligands produced by various pathogens
[29]. Toll-like receptor (TLR) signaling has synergistic and crosstalk effects with CXCR4 and BCR signaling, all of which are mediated by BTK
[30]. In TLR signaling, BTK is required for the downstream activation of NF-κB and interferon regulatory factor-dependent transcription of inflammatory cytokine and interferons due to BTK’s interaction with various proteins, including Myeloid differentiation primary response 88 (MYD88), Toll/Interleukin-1 receptor (TIR), Interleukin-1 receptor-associated kinase 1 (IRAK1) and Toll/Interleukin1 receptor domain-containing adaptor protein (TIRAP)/Myelin and lymphocyte protein (MAL)
[7][28]. Following TLR binding to lipopolysaccharides, most TLRs bind MYD88, initiating a signaling cascade activating TIR, IRAK1 and TIRAP/MAL, leading to BTK activation. BTK’s interplay between TLR and BCR signaling may therefore interconnect the two pathways ()
[31].
Fc Receptor Signaling: Fc receptors are shown to be expressed broadly in hematopoietic lineages and can mediate phagocytosis, release of inflammatory mediators, antibody dependent cell-mediated cytotoxicity and humoral tolerance in different cell types. In plasma cells, FCγRIIB plays an important role in regulating survival and homeostasis
[32]. Activation of FcγRI receptors results in the activation of BTK, STK and PI3K. On the other hand, FCγRIIB receptor activation inhibits BTK through the recruitment of phosphatases. Inhibiting Fc receptor pathways can lead to a reduction in the supportive role of macrophage cells and thus a reduction in vascularization.
BTK Inhibition in MM cells leads to decreases in several key effects of BCR, CXCR, TLR and Fc receptor activation. By blocking BTK and inhibiting all four of these pathways, MM cells causes decreased levels of phosphorylated ERK, AKT, and PLCγ2, yielding decreases in expression of many genes related to oncogenicity including mTORC1, NFκB, ELK1/2, NFAT, MYK and more ().
As previously touched upon, BTK activation through one pathway has important interactions and crosstalk with other pathway molecules. For instance, BTK is activated by the BCR pathway and then can indirectly affect components of the TLR and chemokine signaling pathway, thereby adding complexity to its study
[28]. Due to BTK’s role in activating numerous malignancy related pathways such as NFκB, AKT, mTOR, NFAT and more, the compound appears as an extremely attractive target for a number of blood related malignancies. BTK is particularly suitable as a targeted therapy in MM due to its critical role in the development of B cells within the bone marrow and its effects on numerous components in the BMM ().
 +1 credit
+1 credit