 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | DULCE PERIS-MORENO | + 5009 word(s) | 5009 | 2021-02-08 02:27:56 | | | |
| 2 | Peter Tang | Meta information modification | 5009 | 2021-05-31 04:48:29 | | |
Video Upload Options
Skeletal muscle loss is a detrimental side-effect of numerous chronic diseases that dramatically increases mortality and morbidity. The alteration of protein homeostasis is generally due to increased protein breakdown while, protein synthesis may also be down-regulated. The ubiquitin proteasome system (UPS) is a master regulator of skeletal muscle that impacts muscle contractile properties and metabolism through multiple levers like signaling pathways, contractile apparatus degradation, etc. Among the different actors of the UPS, the E3 ubiquitin ligases specifically target key proteins for either degradation or activity modulation, thus controlling both pro-anabolic or pro-catabolic factors. The atrogenes MuRF1/TRIM63 and MAFbx/Atrogin-1 encode for key E3 ligases that target contractile proteins and key actors of protein synthesis respectively. However, several other E3 ligases are involved upstream in the atrophy program, from signal transduction control to modulation of energy balance. Controlling E3 ligases activity is thus a tempting approach for preserving muscle mass.
1. Introduction
Cachexia is a multifactorial syndrome leading to serious clinical complications with high mortality rates and is present in almost all chronic diseases [1]. Besides inflammation and metabolic modifications, skeletal muscle loss is an important factor of cachexia and limiting muscle wasting is a major challenge for maintaining well-being of patients, the capacity of the organism to fight against diseases and the tolerance of the patients towards challenging therapies like cancer chemotherapies [2].
Muscle homeostasis is mainly driven by the ubiquitin-proteasome system (UPS) that controls signaling pathways, contractile structure, cellular architecture, energy metabolism, protein translation, etc., thus allowing a fine-tuning of skeletal muscle metabolism [3][4][5][6]. The UPS is composed by hundreds of proteins and controls protein fate by ubiquitination, a post-translational modification carried out by the E1, E2, E3 enzymatic cascade (see [7] for a review). Ubiquitin (Ub) is covalently attached to the target proteins thanks to the interactions between Ub conjugating E2 enzymes (35–40 members according to species) and E3 Ub ligases (>600 in human). Another complexity of the UPS resides in the multitude of Ub signals that can be synthesized on the target proteins, from mono-Ub, multiple mono-Ub, or poly-Ub chains with at least eight different topologies. Each type of Ub modification is dedicated to a specific fate for the target protein, the role of some Ub linkages being still obscure. This Ub code can send the target protein for either proteasome or autophagy degradation or for non-proteolytic purposes (addressing, stabilization, activation, etc.) [7]. Furthermore, the multiple possible combinations between a given E3 and several E2s (and vice versa) further increase the potential of the UPS for controlling cellular metabolism.
E3 ligases can be either monomeric or multi-protein complexes and are classified into three families according to their structure and mode of action (recently reviewed [8]). The first class contains 28 members that contain a C-terminal Homologous to E6-Associated Protein C Terminus (HECT) domain that is necessary and sufficient to accept Ub from an E2 enzyme and to transfer it to the substrate, HECT E3 ligases having their own catalytic activity. Their N-terminal domain is involved in the recognition of the substrate. The second class comprises ≈90% of the E3 Ub ligases and are known as Really Interesting New Gene-finger (RING) type. RING domains are defined by eight cysteine and/or histidine residues coordinating four zinc atoms that allow interaction with E2 enzymes. RING-type E3s do not bind Ub, but they serve as a platform for the E2 and the substrate and promote the Ub transfer from the E2 to the substrate. Within multi-protein RING-E3 complexes, also named cullin-containing RING Ligase E3s (CRLs), several families of proteins with motifs involved in protein-protein interactions (e.g., F-box pattern) are responsible for substrate recognition [9]. The third class of E3 ubiquitin ligases are the RING-in-Between-RING (RBR)-type that combine properties of RING- and HECT-type E3s. They utilize an E2-binding RING domain and a second domain (called RING2) that binds Ub before transferring it to substrate [10][11].
Within muscle atrophy, numerous ubiquitinating enzymes are now identified for their involvement in the regulation of both anabolic and catabolic pathways during the atrophy process, notably by being responsible for the degradation of the contractile proteins [12]. The E3 Ub ligases appear to be at the heart of these regulations and some of them may prove to be efficient therapeutic drug strategies with roughly two main approaches: (i) indirect modulation of an E3 ligase by targeting the signals involved in its regulation [13][14][15][16] or (ii) direct inhibition of the E3 ligase [17][18][19]. However, the intertwinement between anabolic and catabolic processes (including the signaling pathways) often renders difficult an indirect modulation of E3 ligases, while direct inhibition strategies is limited by the somehow limited data available on E3 ligases.
2. Signaling Pathways Regulating Skeletal Muscle Mass and Function
Skeletal muscle homeostasis is controlled by numerous signaling pathways (Figure 1) that act either as anabolic or catabolic factors.
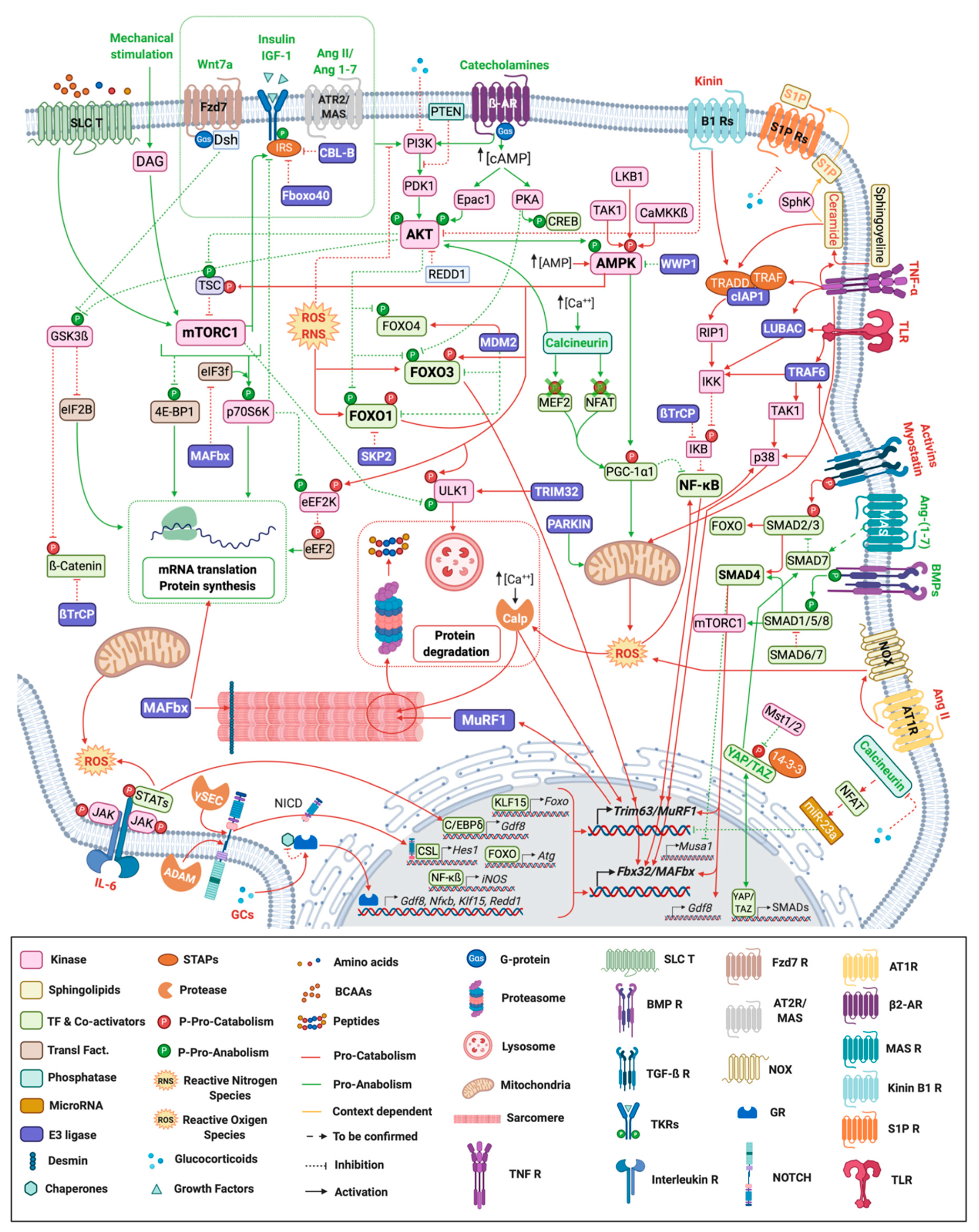
Figure 1. Signaling pathways regulating skeletal muscle mass and function. Myofiber representation of the different signaling pathways controlling skeletal muscle mass and function during atrophy conditions. Ligands and arrows (both with head or perpendicular line) in green denote those signaling pathways and interactions with an anabolic effect whereas the red ones represent catabolic signaling. Orange ligands and arrows stand for pathways with a dual role (context-dependent). ß2-AR: ß-2 Adrenergic Receptor; γ-sec: γ-secretase; Ang: Angiotensin; AT1R: Angiotensin II Type 1 Receptor; AT2R: Angiotensin II Type 2 Receptor; BCAAs: Branched-chain amino acids; BMP R: Bone Morphogenetic Receptor; Calp: Calpain; CSL: CBF1, Suppressor of Hairless, Lag-1; Dsh: Dishevelled; Fzd: Frizzled; GR: Glucocorticoid Receptor; IL-6: Interleukin-6; NCID: Notch Intracellular domain; NOX: NADPH oxidase activator; P: Phosphorylation; S1P: Sphingosine-1-phosphate; SLC T: Solute Carrier Transporter; STAPs: Signal Transducing Adaptor Proteins; TF: Transcription Factors; TGF-ß R: Transforming Growth Factor ß Receptor; TKR: Tyrosine-protein Kinase Receptor; TLR: Toll-like Receptor; TNF R: Tumor Necrosis Factor Receptor; Transl. Fact.: Translational Factors.
3. E3 Ligases Involved in the Regulation of Muscle Atrophy
3.1. E3 Ligases Involved in the Regulation of Anabolic Pathways
3.1.1. The CBL-B and FBXO40 E3 Ubiquitin Ligases Target IRS1 to Degradation in Skeletal Muscle
One strategy to fight against atrophy may be to stimulate the anabolic pathways leading to skeletal muscle hypertrophy. Insulin-like growth factor 1 (IGF1) induces skeletal muscle hypertrophy by activating the IGF1R/PI3K/AKT pathway, a critical mediator and checkpoint being IRS1. Indeed, the effect of IGF1 is time-limited by the phosphorylation of IRS1 by IGF1R and its subsequent ubiquitination and proteasome-mediated degradation.
Different E3 ligases can target IRS1 in different tissues. For example, in embryonic fibroblasts, the CUL7 E3 ligase, containing FBXW8, has been shown to target IRS1 for ubiquitin-dependent degradation [20]. In skeletal muscle, Casitas B-lineage lymphoma-b (CBL-B), a RING E3 ligase, targets IRS1 for degradation and thus impairs muscular trophic signals in response to unloading conditions [21][22][23], which inhibits downstream IGF1 signaling [23] (Figure 2 and Table 1). Accordingly, mice deficient for CBL-B were partly resistant to unloading-induced skeletal muscle atrophy and dysfunction [23]. These results highlight the importance of CBL-B in the process of muscle atrophy in response to unloading.
Table 1. Phenotypes of transgenic mice for genes encoding ubiquitin ligases involved in the control of muscle mass and function.
| Gene Product | E3 Family | Mouse Model | Phenotype | References |
|---|---|---|---|---|
|
E3 ligases regulating the anabolic pathways |
||||
|
CBL-B |
RING |
KO |
Protection from unloading-induced muscle atrophy and dysfunction |
[21] |
|
FBXO40 |
RING |
KD |
Myofibers hypertrophy |
[24] |
|
KO |
Muscle hypertrophy |
|||
|
NEDD4-1 |
HECT |
OX |
Myocardial activation of AKT during I/R |
|
|
KO |
Partially resistant to denervation-induced skeletal muscle atrophy |
[27] |
||
|
E3 ligases regulating the catabolic pathways |
||||
|
TRAF6 |
RING |
m.KO |
Resistance to starvation induced muscle atrophy |
[28] |
|
m.KO |
Resistance to denervation-induced loss of muscle mass and function |
[29] |
||
|
cIAP1 |
RING |
KO |
Limitation of denervation-induced muscle atrophy |
[19] |
|
OX |
Myotube atrophy |
|||
|
WWP1 |
HECT |
KD |
Muscle fiber atrophy |
[30] |
|
TRIM32 |
RING |
KO |
Muscular dystrophy |
[31] |
|
DN |
Muscular dystrophy |
[32] |
||
|
Other E3 ligases involved in the control of muscle mass and function |
||||
|
MuRF1 |
RING |
KO |
Resistance to catabolic-induced muscle atrophy |
[4] |
|
MAFbx |
RING |
KO |
Resistance to catabolic-induced muscle atrophy |
[4] |
|
PARKIN |
RBR |
KO |
Impaired mitochondrial function and muscle atrophy |
[33] |
|
OX |
Increased muscle mass and function in young and old mice |
[34] |
||
|
OX |
Prevention of sepsis-induced muscle atrophy |
[35] |
||
|
SMART/FBXO21 |
RING |
KD |
Resistance to denervation-induced muscle atrophy |
[36] |
|
MUSA1/FBXO30 |
RING |
KD |
Resistance to denervation-induced muscle atrophy |
[37] |
|
FBXL21 |
RING |
HM |
Impaired muscle functions |
[38] |
|
UBR4 |
HECT |
KD |
Muscle hypertrophy |
[39] |
|
UBR5 |
HECT |
KD |
Muscle atrophy |
[40] |
DN, Dominant Negative mutation; HM, Hypomorphic Mutation; I/R, Ischemia/Reperfusion; KD, knock-down mutant; KO, Knock-out mutant; m.KO, skeletal muscle–specific KO mice; OX, overexpressing mutant; PTEN, Phosphatase and tensin homologue.
FBXO40 is a muscle-specific F-box protein [41], component of an SCF (Skp1-Cullin1-F-box protein) E3 ligase complex. Following IRS1 activation, IGF1R phosphorylates IRS1 leading to its ubiquitination by FBXO40 and its degradation by the 26S proteasome, in cultured myotube and in mice [24][42]. FBXO40 expression is decreased in muscles from Limb-girdle muscular dystrophy (LGMD) patients, and up-regulated in mice skeletal muscle following denervation and in chronic kidney disease (CKD) mice model, but not during starvation [41][42]. Accordingly, the knock-down of Fbxo40 resulted in thicker myotubes (20% to 50% increase in diameter) [24] and its deletion in mice also induced muscle hypertrophy during the growth phase, a phase associated with high IGF1 levels [24] (Figure 2 and Table 1).
IRS1 is thus an important checkpoint of the IGF1/PI3K/AKT pathway controlled by at least 2 E3 ligases (CBL-B and FBXO40). Although being an attractive target for fighting against muscle atrophy, the multiple ways for degrading IRS1 may complicate the development of drugs.
3.1.2. NEDD4-1 E3 Ubiquitin Ligase, Friend or Foe?
In muscles undergoing atrophy, NEDD4-1 mRNA levels are elevated upon severe sepsis [43], denervation or unloading [27][44][45]. On the one hand, NEDD4-1 E3 Ub ligase targets phosphatase and tensin homologue (PTEN). PTEN is a redox sensitive phosphatase that negatively regulates the PI3K-AKT signaling pathway, thereby affecting metabolic and cell survival processes. The deletion of PTEN improves muscle mass and function in a mouse model of Duchenne muscular dystrophy [46]. PTEN inhibition may thus also represent a potential therapeutic strategy to maintain muscle function during catabolic situations. The over-expression of NEDD4-1 is sufficient for activating the PI3K/AKT signaling in cardiac muscle, following myocardial ischemia/reperfusion (I/R) [25]. However, the negative regulation of PTEN by NEDD4-1 remains to be confirmed in skeletal muscle, especially since NEDD4-1 has also been shown to promote skeletal muscle atrophy in a denervation model. Indeed, NEDD4-1-KO mice exhibited increased weights and type II muscle fiber cross-sectional areas in denervated gastrocnemius muscle [27]. Moreover, NEDD4-1 also negatively regulates the hypertrophic BMP signaling (Figure 1 and Figure 2). Indeed, NEDD4-1 ubiquitinates phosphorylated-SMAD1 leading to its proteasomal degradation, thereby silencing BMP signaling in C2C12 myoblasts, and conversely the knock-down of Nedd4-1 potentiates BMP signal through upregulation of phospho-SMAD1 [47]. Altogether, the exact function of NEDD4-1 in skeletal muscle is still obscure and needs more work.
3.2. E3 Ubiquitin Ligases Involved in the Regulation of Catabolic Pathways
3.2.1. Regulating the Canonical NF-κB Pathway via the Manipulation of cIAP and TRAF6 E3 Ligases
Among the E3s involved in the regulation of the NF-κB pathway, two promising candidates may be manipulated to limit muscle atrophy, namely cIAP and TRAF6 (Figure 1 and Figure 2). cIAP1 is up-regulated in denervated gastrocnemius muscle, paralleling the upregulation of MAFbx/atrogin-1 and MuRF1/Trim63 mRNA [19]. Mice with genetic ablation of cIAP1 (cIAP1-KO mice) displayed limited denervation-induced atrophy in TA, gastrocnemius and EDL muscles. This was correlated with the blunting of the denervation-induced upregulation of MAFbx/Atrogin-1 and MuRF1/Trim63 [19]. The authors further demonstrated that cIAP1 induced atrophy through the up-regulation of the canonical NF-κB signaling. Conversely, cIAP1 overexpression in myotubes induced atrophy and the strong up-regulation of MAFbx/Atrogin-1 and MuRF1/Trim63 protein expression [19]. The E3 Ub ligase cIAP1 represents thus a potential therapeutic target at least for fighting against denervation-induced muscle atrophy.
TRAF6 is a RING-type Ub ligase that plays an important role during skeletal muscle atrophy. TRAF6 expression is enhanced during starvation or within aged-induced muscle atrophy [28][48][49]. Traf6-KO mice are resistant to skeletal muscle loss (rescue of myofibril degradation, preservation of myofiber size and strength) induced by denervation, cancer cachexia, starvation or Dex and a concomitant suppression of the expression of key regulators of muscle atrophy was observed, including MAFBx/Atrogin-1, MuRF1/TRIM63, p62, Lc3b, Beclin1, Atg12, and Fn14 [28][29][48][49][50]. Moreover, inhibition of Traf6 expression through miR-351 administration in C2C12 myotubes or in denervated mice attenuated Dex-induced muscle atrophy and concomitantly decreased the expression of MAFBx/Atrogin-1 and MuRF1/Trim63 [51][52]. Overexpression of miR-125b targeted Traf6 for degradation and protected skeletal muscle samples from atrophy in starved myotubes or in denervated rat tibialis muscle [53]. The implicated mechanisms involved both direct and indirect effects of TRAF6 on protein breakdown with TRAF6-mediated ubiquitination being required for the optimal activation of JNK, AMPK, FOXO3, and NF-κB catabolic pathway in muscle [54].
In human, gastric cancer patients suffering from cachexia exhibited an upregulation of TRAF6 associated with an upregulation of ubiquitination in the rectus abdominis muscle [55]. Altogether, this highlights the importance for targeting TRAF6 inhibition to counteract muscle atrophy.
3.2.2. WWP1 in the Regulation of Muscle Atrophy
WWP1 is a HECT E3 ligase that is involved in chicken muscular dystrophy. Indeed, a missense mutation in the gene coding WWP1 was identified as the most promising candidate responsible for chicken muscular dystrophy (MD), potentially affecting the E3 function of WWP1 protein [56]. WWP1 was also shown to target the transcription factor KLF15 [30]. In response to glucocorticoids, KLF15 is up-regulated at the mRNA levels [57]. This induction leads to the up-regulation of the E3 ligases MAFbx/Atrogin-1 and MuRF1/Trim63 expression, likely in cooperation with a FOXO transcription factor, while inhibiting the anabolic mTORC1 [57]. Likewise, exogenous KLF15 expression in myotubes and in TA muscle leads to myofiber atrophy [57]. It has recently been shown that KLF15 protein expression was upregulated in skeletal muscle of diabetic mice, without any change in its mRNA expression [30]. This increase correlated with an increase in MAFbx/Atrogin-1, Murf1/Trim63 and Foxo3 genes expression and accordingly, the muscle-specific deletion of Klf15 in this model prevented from diabetes-induced muscle atrophy [30]. The authors identified WWP1 as an E3 ligase targeting KLF15 and showed that knocking-down WWP1 in both C2C12 myotubes and in tibialis anterior muscles increased MuRF1/Trim63 and MAFbx/Atrogin-1 expression and induced atrophy [30] (Figure 2). WWP1 E3 ligase is indeed induced by high glucose conditions in myotubes [58]. Conversely, in high glucose conditions, WWP1 has also been implicated in the down-regulation of AMPKα2 protein levels [58]. The authors have shown that WWP1 interacted with AMPKα2 leading to a proteasome-dependent decrease of AMPKα2 in myotubes; however, direct ubiquitination was not addressed [58]. WWP1 may thus control muscle mass through a direct action on AMPK, a known modulator of FOXO3a, MuRF1/TRIM63 and MAFbx/Atrogin-1 [59].
3.2.3. TRIM32 in the Regulation of Autophagy
TRIM32 is a RING E3 Ub ligase whose mutation is responsible for the development of limb girdle dystrophy 2H (LGMD2H) [60]. Several substrates have been identified for TRIM32 in non-muscle cells, including cell cycle regulators (c-Myc, MYCN, p53), the cell growth and transformation factor ABI2 and PIASY (a SUMO E3 ligase). TRIM32 is also involved in the targeting of factors influencing myogenesis (NDRG2 and TRIM72) that regulate muscle satellite cells renewal and differentiation [61]. While initially postulated to promote muscle atrophy, TRIM32 is in fact a master regulator of myogenesis during recovery situations [61]. Indeed, the dystrophic phenotype of TRIM32 mutations appeared to be largely due to impaired myogenesis [61][62][63].
More recently, TRIM32 was implicated in the early events leading to autophagy. Indeed, TRIM32 targets ULK1, a Ser/Thr protein kinase (Figure 1 and Figure 2). ULK1 is an upstream regulator of autophagy rapidly activated to ensure a rapid response to stress conditions [64]. The authors showed that TRIM32 deficiency was directly responsible for autophagy defects both in cultured cells and in mice treated with Dex. The mechanisms by which TRIM32 controls the activation of autophagy through ULK1 involves its binding to AMBRA1, a positive regulator of autophagy [64]. AMBRA1 is a pivotal factor able to bind several E3 ligases during the course of the autophagy process. In presence of AMBRA1, TRIM32 binds to ULK1, synthesizes unanchored K63 Ub chains that activate ULK1 kinase activity, thus promoting autophagy. The role of TRIM32 during the autophagy process is not limited to ULK1 as p62, an important autophagy receptor [65], is also a TRIM32 substrate. p62 activity is modulated by multi mono-Ub catalyzed by TRIM32 and loss of function of TRIM32 largely abolished autophagy [66]. Altogether, TRIM32 appears as a master regulator of muscle renewal through the initiation of autophagy.
3.2.4. FOXO Transcription Factors Are Regulated by MDM2 and SKP2 E3 Ubiquitin Ligases
Alternatively to phosphorylation, FOXO can be regulated by acetylation/deacetylation, methylation and ubiquitination to modulate its activity, localization as well as degradation [67][68][69].
Ubiquitination modulate FOXO activity by either mono- or polyubiquitination through MDM2 and SKP2 E3 Ub ligases (Figure 1 and Figure 2). MDM2 is the enzyme responsible of a single addition of an ubiquitin moiety to FOXOs, specifically to FOXO4, thus allowing its nuclear localization and transcriptional activation [70][71]. Mono-Ub of FOXO4 is observed under oxidative stress conditions and can be counteracted by deubiquitinating enzymes such as ubiquitin-specific protease (USP7). Importantly, ubiquitination mediated by MDM2 is context specific and upon growth factor stimulation can induce FOXO1 and 3 degradation [70]. In addition, interaction between FOXOs and SKP2, a subunit of the SKP/cullin 1/F-box protein E3 ligase leads to proteasomal degradation of FOXO1 in the cytosol [71].
Combined with the other posttranslational modifications, ubiquitination allows FOXOs to integrate information arising from insulin, growth factors, cytokines, and oxidative stress and to control downstream signaling. Interestingly, FOXO TFs have systematically been envisioned as crucial drivers of catabolic pathways during muscle wasting. Nonetheless, recent work showed that FOXO1 and 3a participate to skeletal muscle adaptation upon exercise thus adding a new of FOXOs in the control of muscle cell homeostasis [72][73][74][75].
3.3. E3 Ubiquitin Ligases Involved in the Regulation of Muscle Mass and Function
3.3.1. MuRF1/TRIM63
Muscle-specific RING finger protein 1 (MuRF1), also named TRIM63, is a RING-type E3 ligase and a founding member of the so-called “atrogenes” (see [6] for a recent review). MuRF1/TRIM63 is a master regulator of skeletal muscle atrophy development occurring in numerous catabolic conditions and MuRF1/Trim63 mRNA appeared to be upregulated in more than 25 atrophying situations [6] (Figure 1 and Figure 2). Mice deleted for MuRF1/TRIM63 (MuRF1-KO mice) were partially resistant (preservation of muscle mass and structure) to skeletal muscle atrophy induced by denervation [4], hindlimb suspension [4][76], glucocorticoid [77], amino acid deprivation [78], and acute lung injury [79]. MuRF1/TRIM63 is responsible for the coordinated breakdown of both thick and thin filaments occurring during catabolic states in skeletal muscle, targeting to degradation the main proteins of the contractile apparatus: myosin heavy chains (MHC) [80], alpha-actin [81], troponin I [82], TCAP/telethonin [83]. During denervation and starvation, MuRF1/TRIM63 has also been involved in the degradation of acetylcholine receptor (CHRN), the major postsynaptic ion channel of the neuromuscular junction. This degradation is mediated by the activation of selective autophagy and degradation of CHRN, likely via the degradation of BIF-1 (Bax interacting factor 1)/EndoB1 (EndophilinB1) and/or SQTM1/p62 (sequestosome-1) [84][85].
While numerous studies have promoted a major role of MuRF1/TRIM63 in the development of skeletal muscle atrophy during catabolic states, in the heart, the analyses of MuRF1 mutants have highlighted a beneficial cardioprotective role [86]. These opposites roles in both kind of muscles imply the development of skeletal muscle-specific drugs to inhibit MuRF1/TRIM63. Moreover, one should also take into account that MuRF1/TRIM63 has two homologs, MuRF2 and MuRF3 that share some redundant functions and could replace its role [12].
3.3.2. MAFbx/Atrogin-1/FBXO32
The multimeric E3 ligase MAFbx/atrogin-1/FBXO32 is another founding member of the atrogene family ([6] for a recent review) crucial for the development of muscle atrophy. Interestingly, nearly all catabolic situations induce an overexpression of both MAFbx/Atrogin-1 and MuRF1/TRIM63, which are controlled by the same TFs (FOXO1/FOXO3a, NF-κB, C/EBP β, Smad 3, etc.) and the same signaling pathways [87] (Figure 1 and Figure 2).
In contrast with MuRF1/TRIM63 that targets directly the contractile proteins for their degradation (α-actin, MHC, etc. [80][81][82][83], MAFbx appeared to target pro-anabolic factors like MyoD, myogenin or eIF3f [88][89][90]. MyoD is a muscle-specific transcription factor that plays crucial roles during cell cycle and muscle differentiation [91]. The eukaryotic initiation factor 3 subunit f (eIF3f) is a pivotal element of protein synthesis and its control by MAFbx allows the latter to master the anabolic processes [88]. While a putative role of MAFbx/Atrogin-1 on sarcomeric proteins was hypothesized using an indirect approach, this has never been confirmed [92]. By contrast, the authors found that desmin, a main component of the intermediate filaments, physically interacted with MAFbx and was degraded in myostatin-treated cultured C2C12 myotubes.
As MAFbx/Atrogin-1 and MuRF1/TRIM63 are controlled by similar signaling pathways, the strategies for the upstream control of MuRF1/Trim63 expression are generally also valid for MAFbx/Atrogin-1 (Table 2). By contrast with MuRF1/TRIM63, no direct inhibitor of MAFbx/Atrogin-1 has been described so far but general strategies, like targeting the interface responsible for substrate recognition or impeding the assembly of the F-box (i.e., the subunit recognizing the substrates) into the SCF complex, may prove to be efficient.
Altogether, controlling concomitantly MAFbx/Atrogin-1 and MuRF1/TRIM63 E3 ligases allows skeletal muscle cells to both increase the degradation of the contractile apparatus and to depress the protein synthesis machinery, which allows a tight regulation of protein homeostasis.
3.3.3. PARKIN Controls Muscle Mass through the Maintenance of Mitochondrial Homeostasis
PARKIN is an E3 ubiquitin ligase implicated in the regulation of mitophagy, a quality control process in which defective mitochondria are degraded. Mitochondrial quality control through both mitochondria turnover and dynamic plays an essential role in the maintenance of muscle mass (see [93] for a review). During mitophagy, PARKIN ubiquitinates several outer mitochondrial membrane proteins leading to subsequent autophagosomal engulfment and lysosomal degradation (Figure 1 and Figure 2).
This role of PARKIN has been emphasized in rodent models or in humans where a deregulation of PARKIN mRNA and/or protein expression prevailed in response to catabolic or anabolic situations. An accumulation of PARKIN protein prevailed during: (i) muscle wasting situations such as chronic kidney disease [94], chronic obstructive pulmonary disease (COPD) [95], physical inactivity [96][97] and (ii) upon exercise training [98][99]. Conversely, PARKIN mRNA or protein levels decreases in skeletal muscles from some elderly populations, perhaps related to the loss of muscle mass and poor physical function, e.g., physically inactive frail older women [100][101] or gastric cancer patients with cachexia [102].
In the last two years many studies using loss/gain of function models have provided insight on the role of PARKIN in skeletal muscle. Loss of function mouse models pointed out the essential role of PARKIN in basal conditions for the maintenance of (i) mitochondrial function [103][104] and (ii) skeletal muscle mass and normal contractile function [33][104]. Such studies also reported that PARKIN helps to resist to some drug-induced muscle damages [105] and is required for exercise-induced mitophagy flux and for the accumulation of functional mitochondria following muscle adaptations to training [103]. In addition, these loss-of-function studies also highlighted that PARKIN-mediated mitochondrial clearance contributes to proteasome activation during denervation in atrophied slow-twitch muscles [106]. On the flip side, gain-of-function studies showed that PARKIN overexpression in mice: (i) attenuates the ageing-related and the sepsis-induced muscle wasting and causes hypertrophy in adult skeletal muscle, (ii) increases mitochondrial content and enzymatic activities and (iii) protects from ageing-related increases of oxidative stress markers, fibrosis and apoptosis [34][35]. It is very likely that this role of PARKIN in controlling muscle mass has been evolutionary conserved. Indeed, similar observations were also reported in the fruit fly model: Parkin deficiency in Drosophila leads to severe degeneration of the flight muscles with accumulation of swollen mitochondria [107] whereas Parkin overexpression promotes mitophagy in older muscles and extend lifespan.
Together, these studies clearly indicate that PARKIN is an important player in the control of muscle mass through its role in the maintenance of mitochondrial homeostasis. This makes it a potential therapeutic target of interest for preserving muscle mass or fighting against atrophy. Nevertheless, the regulation of PARKIN can be very different according to the physiological or pathological situation or during ageing. Further investigations should enable defining how this actor could be a target of interest according to the population considered.
3.3.4. MUSA1/FBXO30
FBXO30, also called muscle ubiquitin ligase of the SCF complex in atrophy-1 (MUSA1), is a FBOX protein forming an SCF complex with SKP1, Cullin1 and ROC1 [37]. Proteins targeted by MUSA1 remain undefined, but its inhibition in denervated muscles reduces remarkably muscle atrophy, and reverts almost completely the strong atrophic phenotype of Smad4-KO mice [37] (Figure 1 and Figure 2). In muscle, Musa1 expression is upregulated in atrophic mice muscle undergoing CKD [108] or sepsis [109].
3.3.5. FBXL21
Very recently, a new E3 ubiquitin ligase involved in muscle function control has emerged, FBXL21 [38]. FBXL21 forms an SCF E3 ligase complex and was first identified as clock-controlled E3 ligase modulating circadian periodicity via subcellular cryptochrome degradation [110]. Accordingly, in mice, the Psttm mutation, corresponding to a hypomorphic mutation of FBXL21 with reduced FBXL21 activity, caused circadian period shortening [110]. Further studies of these mice revealed that they also displayed skeletal muscle deficiencies with a decrease in fiber CSA (gastrocnemius) and impaired exercise tolerance and grip strength for both forelimbs and hindlimbs [38]. The authors nicely demonstrated the circadian degradation of the cytosolic TCAP/Telethonin by FBXL21 (Figure 2), under the control of GSK-3β. They reported that GSK-3β phosphorylated both FBXL21 and TCAP leading to FBXL21-CULLIN1 complex formation and phosphodegron-dependent TCAP turnover.
3.3.6. Ubiquitin Ring-Type E3 Ligases (UBR)
Ubiquitin Ring-type (UBR, also referred to as E3α) proteins are RING finger E3 ligases that compose a 7-member family and that mainly recognize their substrate through the N-end rule pathway [111]. A first member, UBR2/E3alpha-II, has been shown to be significantly induced in skeletal muscle, in two different animal models of cancer cachexia, at the onset and during the progression of muscle wasting [112]. However, its exact function and importance in skeletal muscle maintain during catabolic states have not been further studied. UBR4 is overexpressed in the skeletal from fasted mice and genetic ablation of UBR4 preserves muscle mass in tumor-bearing mice [39] (Table 1). Intriguingly, the protection of UBR4 knockout against tumor-induced atrophy was limited to type IIA fibers. In contrast, UBR5 has been implicated in muscle hypertrophy [113] and reported to be at least partially associated to the proteasome [114]. Recently several members of the UPS have been described as UBR5 substrates, which included an E2 (UBE2B, an abundant muscle E2), several E3 ligases, proteins involved in chromatin remodeling, etc. [39]. As the main UBR4 targets are positive regulators of muscle growth, the authors concluded that UBR4 acts as a negative regulator of muscle hypertrophy.
3.3.7. FBXO21/SMART
FBXO21/SMART forms an SCF complex with Skp1, Cullin1 and Roc1, in skeletal muscle and has been shown to promote atrophy during denervation [36]. Indeed, the authors showed that FBXO21/SMART upregulation was required for atrophy while, knock-down in TA muscle protected denervated muscles from atrophy (Table 1), probably due to a global reduction of protein ubiquitination [36]. FBXO21/SMART might therefore be a new critical E3 to target to limit skeletal muscle atrophy. Further work should determine whether this E3 is crucial for the development of atrophy in other catabolic conditions and what are the mechanisms involved.
3.4. Promising E3 Ubiquitin Ligases Regulating Muscle Mass and Function
Other E3 ubiquitin ligases are also promising putative targets for maintaining muscle mass and function, if we rely on what has been published in other organs or organisms. For example, the SIAH-1 RING E3 ligase has been identified in the same RNAi screen that UBR4, performed to identify ubiquitin-related enzymes that regulate myofiber size, using the fruit fly Drosophila [39]. In Drosophila, SIAH1 knock-down led to muscular hypertrophy while its overexpression led to atrophy [39]. It is noteworthy that, in space flown rats, SIAH1 mRNA expression has been shown upregulated suggesting also a putative role during this process in mammals [22]. However, in mammals two isoforms, SIAH1 and SIAH2, are expressed in muscle and could share redundant functions [39].
SMURF1, an HECT ubiquitin ligase interacts with SMAD1 and SMAD5 (BMP pathway) and SMAD4 in a certain context, leading them all to proteasomal degradation in vitro [115]. Moreover, it can degrade the main TGF-β receptor through an indirect recruitment to the receptor by SMAD7, leading to the receptor degradation [116]. In, COPD leading to muscle atrophy, TGF-β signaling is abnormally up-regulated and this, is negatively correlated to SMURF1 expression. This highlights that the inhibitory effect of SMURF1 over TGF-β is needed for muscle homeostasis [117].
The C terminus of Hsc70-interacting protein (STUB1/CHIP) serves as an E3 ubiquitin ligase. This E3 plays a dual role in BMP/TGF signaling. Overexpression of CHIP inhibits TGF-β luciferase reporter through the ubiquitination and degradation of SMAD3, and conversely silencing it leads to increase the signal transduction in HEK293T cells [118]. In cellulo experiments showed that CHIP mediates as well SMAD1-5 poly-ubiquitination, and subsequent degradation to terminate BMP signaling [119]. In muscle, CHIP is highly expressed. For instance, Chip−/− mice at 6 months shows muscle morphological changes consistent with increased sarcoplasmic reticulum compartments in quadriceps muscle and gastrocnemius, resulting in damages and fiber switch composition [120]. From our knowledge, no studies have shown the implication of CHIP in TGF/BMP signaling-mediated muscle atrophy.
TRIM62 belongs to the TRIM/RBCC family. This enzyme acts as a negative regulator of TGF-β signaling by binding to SMAD3 and promoting its ubiquitination and degradation, resulting in a decrease of TGF-β/SMAD3 target genes in HEK and human mammary epithelial cells [121]. TRIM62 is increased in the skeletal muscle of ICUAW patients (Intensive care unit-acquired weakness), a devastating illness characterized by loss of muscle mass [122]. In this context, the authors proposed TRIM62 contribution in inflammation-induced muscle atrophy through IL-6 pathway. Indeed, Trim62-KD inhibited LPS-induced IL-6 expression in C2C12 cells [122].
TRIM72/MG53 is a muscle-specific E3 ligase, also called mitsugumin 53, specifically expressed in the plasma membrane of skeletal muscle, and has a critical role in membrane repair. Membrane repair deficiency causes muscle cell death, injury, and dystrophy. Accordingly, the overexpression of human TRIM72 in a hamster model of genetic muscular dystrophy protects skeletal muscle damage through enhancement of membrane repair [123]. Similarly, short-term TRIM72 injection ameliorates the underlying defects in dysferlin-deficient muscle by increasing sarcolemma membrane integrity [124] while Trim72−/− mice develop significant skeletal muscle myopathy and cardiovascular defects due to defective sarcolemma repair [125].
References
- Von Haehling, S.; Anker, M.S.; Anker, S.D. Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: Facts and numbers update 2016. J. Cachexia Sarcopenia Muscle 2016, 7, 507–509.
- Penna, F.; Ballarò, R.; Beltrà, M.; De Lucia, S.; García Castillo, L.; Costelli, P. The Skeletal Muscle as an Active Player against Cancer Cachexia. Front. Physiol. 2019, 10, 41.
- Blondelle, J.; Biju, A.; Lange, S. The Role of Cullin-RING Ligases in Striated Muscle Development, Function, and Disease. Int. J. Mol. Sci. 2020, 21, 7936.
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708.
- Polge, C.; Attaix, D.; Taillandier, D. Role of E2-Ub-conjugating enzymes during skeletal muscle atrophy. Front. Physiol. 2015, 6, 59.
- Taillandier, D.; Polge, C. Skeletal muscle atrogenes: From rodent models to human pathologies. Biochimie 2019, 166, 251–269.
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends. Biochem. Sci. 2017, 42, 873–886.
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157.
- Weissman, A.M. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell. Biol. 2001, 2, 169–178.
- Walden, H.; Rittinger, K. RBR ligase–mediated ubiquitin transfer: A tale with many twists and turns. Nat. Struct. Mol. Cell. Biol. 2018, 25, 440–445.
- Dove, K.K.; Klevit, R.E. RING-between-RING E3 Ligases: Emerging Themes amid the Variations. J. Mol. Biol. 2017, 429, 3363–3375.
- Peris-Moreno, D.; Taillandier, D.; Polge, C. MuRF1/TRIM63, Master Regulator of Muscle Mass. Int. J. Mol. Sci. 2020, 21, 6663.
- Frost, R.A.; Nystrom, G.J.; Jefferson, L.S.; Lang, C.H. Hormone, cytokine, and nutritional regulation of sepsis-induced increases in atrogin-1 and MuRF1 in skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2007, 292, E501–E512.
- Gopinath, S.D. Inhibition of stat3 signaling ameliorates atrophy of the soleus muscles in mice lacking the vitamin D receptor. Skelet. Muscle 2017, 7, 1–17.
- Kline, W.O.; Panaro, F.J.; Yang, H.; Bodine, S.C. Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J. Appl. Physiol. 2007, 102, 740–747.
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of Cancer Cachexia and Muscle Wasting by ActRIIB Antagonism Leads to Prolonged Survival. Cell 2010, 142, 531–543.
- Adams, V.; Gußen, V.; Zozulya, S.; Cruz, A.; Moriscot, A.; Linke, A.; Labeit, S. Small-Molecule Chemical Knockdown of MuRF1 in Melanoma Bearing Mice Attenuates Tumor Cachexia Associated Myopathy. Cells 2020, 9, 2272.
- Adams, V.; Bowen, T.S.; Werner, S.; Barthel, P.; Amberger, C.; Konzer, A.; Graumann, J.; Sehr, P.; Lewis, J.; Provaznik, J.; et al. Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J. Cachexia Sarcopenia Muscle 2019, 10, 1102–1115.
- Lala-Tabbert, N.; Lejmi-Mrad, R.; Timusk, K.; Fukano, M.; Holbrook, J.; St-Jean, M.; LaCasse, E.C.; Korneluk, R.G. Targeted ablation of the cellular inhibitor of apoptosis 1 (cIAP1) attenuates denervation-induced skeletal muscle atrophy. Skelet. Muscle 2019, 9, 1–13.
- Xu, X.; Sarikas, A.; Dias-Santagata, D.C.; Dolios, G.; Lafontant, P.J.; Tsai, S.-C.; Zhu, W.; Nakajima, H.; Nakajima, H.-O.; Field, L.J.; et al. The CUL7 E3 Ubiquitin Ligase Targets Insulin Receptor Substrate 1 for Ubiquitin-Dependent Degradation. Mol. Cell. 2008, 30, 403–414.
- Nakao, R.; Hirasaka, K.; Goto, J.; Ishidoh, K.; Yamada, C.; Ohno, A.; Okumura, Y.; Nonaka, I.; Yasutomo, K.; Baldwin, K.M.; et al. Ubiquitin Ligase Cbl-b Is a Negative Regulator for Insulin-Like Growth Factor 1 Signaling during Muscle Atrophy Caused by Unloading. Mol. Cell. Biol. 2009, 29, 4798–4811.
- Nikawa, T.; Ishidoh, K.; Hirasaka, K.; Ishihara, I.; Ikemoto, M.; Kano, M.; Kominami, E.; Nonaka, I.; Ogawa, T.; Adams, G.R.; et al. Skeletal muscle gene expression in space-flown rats. FASEB J. 2004, 18, 522–524.
- Uchida, T.; Sakashita, Y.; Kitahata, K.; Yamashita, Y.; Tomida, C.; Kimori, Y.; Komatsu, A.; Hirasaka, K.; Ohno, A.; Nakao, R.; et al. Reactive oxygen species upregulate expression of muscle atrophy-associated ubiquitin ligase Cbl-b in rat L6 skeletal muscle cells. Am. J. Physiol.-Cell Physiol. 2018, 314, C721–C731.
- Shi, J.; Luo, L.; Eash, J.; Ibebunjo, C.; Glass, D.J. The SCF-Fbxo40 Complex Induces IRS1 Ubiquitination in Skeletal Muscle, Limiting IGF1 Signaling. Dev. Cell. 2011, 21, 835–847.
- Hu, W.; Zhang, P.; Gu, J.; Yu, Q.; Zhang, D. NEDD4-1 protects against ischaemia/reperfusion-induced cardiomyocyte apoptosis via the PI3K/Akt pathway. Apoptosis 2017, 22, 437–448.
- Wang, X.; Trotman, L.C.; Koppie, T.; Alimonti, A.; Chen, Z.; Gao, Z.; Wang, J.; Erdjument-Bromage, H.; Tempst, P.; Cordon-Cardo, C.; et al. NEDD4-1 Is a Proto-Oncogenic Ubiquitin Ligase for PTEN. Cell 2007, 128, 129–139.
- Nagpal, P.; Plant, P.J.; Correa, J.; Bain, A.; Takeda, M.; Kawabe, H.; Rotin, D.; Bain, J.R.; Batt, J.A.E. The Ubiquitin Ligase Nedd4-1 Participates in Denervation-Induced Skeletal Muscle Atrophy in Mice. PLoS ONE 2012, 7, e46427.
- Paul, P.K.; Bhatnagar, S.; Mishra, V.; Srivastava, S.; Darnay, B.G.; Choi, Y.; Kumar, A. The E3 Ubiquitin Ligase TRAF6 Intercedes in Starvation-Induced Skeletal Muscle Atrophy through Multiple Mechanisms. Mol. Cell. Biol. 2012, 32, 1248–1259.
- Paul, P.K.; Gupta, S.K.; Bhatnagar, S.; Panguluri, S.K.; Darnay, B.G.; Choi, Y.; Kumar, A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J. Cell Biol. 2010, 191, 1395–1411.
- Hirata, Y.; Nomura, K.; Senga, Y.; Okada, Y.; Kobayashi, K.; Okamoto, S.; Minokoshi, Y.; Imamura, M.; Takeda, S.; Hosooka, T.; et al. Hyperglycemia induces skeletal muscle atrophy via a WWP1/KLF15 axis. JCI Insight 2019, 4, e124952.
- Kudryashova, E.; Wu, J.; Havton, L.A.; Spencer, M.J. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 2009, 18, 1353–1367.
- Kudryashova, E.; Struyk, A.; Mokhonova, E.; Cannon, S.C.; Spencer, M.J. The common missense mutation D489N in TRIM32 causing limb girdle muscular dystrophy 2H leads to loss of the mutated protein in knock-in mice resulting in a Trim32-null phenotype. Hum. Mol. Genet. 2011, 20, 3925–3932.
- Peker, N.; Donipadi, V.; Sharma, M.; McFarlane, C.; Kambadur, R. Loss of Parkin impairs mitochondrial function and leads to muscle atrophy. Am. J. Physiol.-Cell Physiol. 2018, 315, C164–C185.
- Leduc-Gaudet, J.P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G. Parkin overexpression protects from ageing-related loss of muscle mass and strength. J. Physiol. 2019, 597, 1975–1991.
- Leduc-Gaudet, J.-P.; Mayaki, D.; Reynaud, O.; Broering, F.E.; Chaffer, T.J.; Hussain, S.N.A.; Gouspillou, G. Parkin Overexpression Attenuates Sepsis-Induced Muscle Wasting. Cells 2020, 9, 1454.
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin–proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670.
- Sartori, R.; Schirwis, E.; Blaauw, B.; Bortolanza, S.; Zhao, J.; Enzo, E.; Stantzou, A.; Mouisel, E.; Toniolo, L.; Ferry, A.; et al. BMP signaling controls muscle mass. Nat. Genet. 2013, 45, 1309–1321.
- Wirianto, M.; Yang, J.; Kim, E.; Gao, S.; Paudel, K.R.; Choi, J.M.; Choe, J.; Gloston, G.F.; Ademoji, P.; Parakramaweera, R.; et al. The GSK-3β-FBXL21 Axis Contributes to Circadian TCAP Degradation and Skeletal Muscle Function. Cell Rep. 2020, 32, 108140.
- Hunt, L.C.; Stover, J.; Haugen, B.; Shaw, T.I.; Li, Y.; Pagala, V.R.; Finkelstein, D.; Berton, E.R.; Fan, Y.; Labelle, M.; et al. A Key Role for the Ubiquitin Ligase UBR4 in Myofiber Hypertrophy in Drosophila and Mice. Cell Rep. 2019, 28, 1268–1281.e6.
- Hughes, D.C.; Turner, D.C.; Baehr, L.M.; Seaborne, R.A.; Viggars, M.; Jarvis, J.C.; Gorski, P.P.; Stewart, C.E.; Owens, D.J.; Bodine, S.C.; et al. Knockdown of the E3 Ubiquitin ligase UBR5 and its role in skeletal muscle anabolism. Am. J. Physiol.-Cell Physiol. 2020, 320, C45–C56.
- Ye, J.; Zhang, Y.; Xu, J.; Zhang, Q.; Zhu, D. FBXO40, a gene encoding a novel muscle-specific F-box protein, is upregulated in denervation-related muscle atrophy. Gene 2007, 404, 53–60.
- Zhang, L.; Chen, Z.; Wang, Y.; Tweardy, D.J.; Mitch, W.E. Stat3 activation induces insulin resistance via a muscle-specific E3 ubiquitin ligase Fbxo40. Am. J. Physiol.-Endocrinol. Metab. 2020, 318, E625–E635.
- Stana, F.; Vujovic, M.; Mayaki, D.; Leduc-Gaudet, J.P.; Leblanc, P.; Huck, L.; Hussain, S.N.A. Differential Regulation of the Autophagy and Proteasome Pathways in Skeletal Muscles in Sepsis. Crit. Care Med. 2017, 45, e971–e979.
- Batt, J.; Bain, J.; Goncalves, J.; Michalski, B.; Plant, P.; Fahnestock, M.; Woodgett, J. Differential gene expression profiling of short and long term denervated muscle. FASEB J. 2006, 20, 115–117.
- Koncarevic, A.; Jackman, R.W.; Kandarian, S.C. The ubiquitin-protein ligase Nedd4 targets Notch1 in skeletal muscle and distinguishes the subset of atrophies caused by reduced muscle tension. FASEB J. 2007, 21, 427–437.
- Yue, F.; Song, C.; Huang, D.; Narayanan, N.; Qiu, J.; Jia, Z.; Yuan, Z.; Oprescus, S.N.; Roseguini, B.T.; Deng, M.; et al. PTEN Inhibition Ameliorates Muscle Degeneration and Improves Muscle Function in a Mouse Model of Duchenne Muscular Dystrophy. Mol. Therap. 2020.
- Kim, B.G.; Lee, J.H.; Yasuda, J.; Ryoo, H.M.; Cho, J.Y. Phospho-Smad1 modulation by nedd4 e3 ligase in BMP/TGF-β signaling. J. Bone Min. Res. 2011, 26, 1411–1424.
- Li, J.; Yi, X.; Yao, Z.; Chakkalakal, J.V.; Xing, L.; Boyce, B.F. TNF Receptor-Associated Factor 6 Mediates TNFα-Induced Skeletal Muscle Atrophy in Mice During Aging. J. Bone Min. Res. 2020, 35, 1535–1548.
- Sun, H.; Gong, Y.; Qiu, J.; Chen, Y.; Ding, F.; Zhao, Q. TRAF6 inhibition rescues dexamethasone-induced muscle atrophy. Int. J. Mol. Sci. 2014, 15, 11126–11141.
- Sun, H.; Qiu, J.; Chen, Y.; Yu, M.; Ding, F.; Gu, X. Proteomic and bioinformatic analysis of differentially expressed proteins in denervated skeletal muscle. Int. J. Mol. Med. 2014, 33, 1586–1596.
- He, Q.; Qiu, J.; Dai, M.; Fang, Q.; Sun, X.; Gong, Y.; Ding, F.; Sun, H. MicroRNA-351 inhibits denervation-induced muscle atrophy by targeting TRAF6. Exp. Therap. Med. 2016, 12, 4029–4034.
- Qiu, J.; Wang, L.; Wang, Y.; Zhang, Q.; Ma, W.; Fang, Q.; Sun, H.; Ding, F. MicroRNA351 targeting TRAF6 alleviates dexamethasone-induced myotube atrophy. J. Thorac. Dis. 2018, 10, 6238–6246.
- Qiu, J.; Zhu, J.; Zhang, R.; Liang, W.; Ma, W.; Zhang, Q.; Huang, Z.; Ding, F.; Sun, H. miR-125b-5p targeting TRAF6 relieves skeletal muscle atrophy induced by fasting or denervation. Ann. Transl. Med. 2019, 7, 456.
- Paul, P.K.; Kumar, A. TRAF6 coordinates the activation of autophagy and ubiquitin-proteasome systems in atrophying skeletal muscle. Autophagy 2011, 7, 555–556.
- Sun, Y.S.; Ye, Z.Y.; Qian, Z.Y.; Xu, X.D.; Hu, J.F. Expression of TRAF6 and ubiquitin mRNA in skeletal muscle of gastric cancer patients. J. Exp. Clin. Cancer Res. 2012, 31, 1–5.
- Imamura, M.; Nakamura, A.; Mannen, H.; Takeda, S. Characterization of WWP1 protein expression in skeletal muscle of muscular dystrophy chickens. J. Biochem. 2016, 159, 171–179.
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.I.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 2011, 13, 170–182.
- Lee, J.O.; Lee, S.K.; Kim, N.; Kim, J.H.; You, G.Y.; Moon, J.W.; Jie, S.; Kim, S.J.; Lee, Y.W.; Kang, H.J.; et al. E3 ubiquitin ligase, WWP1, interacts with AMPKα2 and down-regulates its expression in skeletal muscle C2C12 cells. J. Biol. Chem. 2013, 288, 4673–4680.
- Thomson, D.M. The Role of AMPK in the Regulation of Skeletal Muscle Size, Hypertrophy, and Regeneration. Int. J. Mol. Sci. 2018, 19, 3125.
- Frosk, P.; Weiler, T.; Nylen, E.; Sudha, T.; Greenberg, C.R.; Morgan, K.; Fujiwara, T.M.; Wrogemann, K. Limb-Girdle Muscular Dystrophy Type 2H Associated with Mutation in TRIM32, a Putative E3-Ubiquitin–Ligase Gene. Am. J. Hum. Genet. 2002, 70, 663–672.
- Mokhonova, E.I.; Avliyakulov, N.K.; Kramerova, I.; Kudryashova, E.; Haykinson, M.J.; Spencer, M.J. The E3 ubiquitin ligase TRIM32 regulates myoblast proliferation by controlling turnover of NDRG2. Hum. Mol. Genet. 2015, 24, 2873–2883.
- Kudryashova, E.; Kramerova, I.; Spencer, M.J. Satellite cell senescence underlies myopathy in a mouse model of limb-girdle muscular dystrophy 2H. J. Clin. Investig. 2012, 122, 1764–1776.
- Servián-Morilla, E.; Cabrera-Serrano, M.; Rivas-Infante, E.; Carvajal, A.; Lamont, P.J.; Pelayo-Negro, A.L.; Ravenscroft, G.; Junckerstorff, R.; Dyke, J.M.; Fletcher, S.; et al. Altered myogenesis and premature senescence underlie human TRIM32-related myopathy. Acta Neuropathol. Commun. 2019, 7, 30.
- Di Rienzo, M.; Antonioli, M.; Fusco, C.; Liu, Y.; Mari, M.; Orhon, I.; Refolo, G.; Germani, F.; Corazzari, M.; Romagnoli, A.; et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci. Adv. 2019.
- Peng, H.; Yang, J.; Li, G.; You, Q.; Han, W.; Li, T.; Gao, D.; Xie, X.; Lee, B.-H.; Du, J.; et al. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res. 2017, 27, 657–674.
- Overå, K.S.; Garcia-Garcia, J.; Bhujabal, Z.; Jain, A.; Øvervatn, A.; Larsen, K.B.; Johansen, T.; Lamark, T.; Sjøttem, E. TRIM32, but not its muscular dystrophy-associated mutant, positively regulates and is targeted to autophagic degradation by p62/SQSTM1. J. Cell Sci. 2019, 132.
- Alamdari, N.; Aversa, Z.; Castillero, E.; Hasselgren, P.-O. Acetylation and deacetylation—Novel factors in muscle wasting. Metabolism 2013, 62, 1–11.
- Bertaggia, E.; Coletto, L.; Sandri, M. Posttranslational modifications control FoxO3 activity during denervation. Am. J. Physiol.-Cell Physiol. 2012, 302, C587–C596.
- Kim, H.; Kang, J.-S.; Jeong, H.-J. Arginine methylation as a key post-translational modification in skeletal muscle homeostasis: A review. Precis. Future Med. 2019, 3, 139–145.
- Brown, A.K.; Webb, A.E. Regulation of FOXO Factors in Mammalian Cells. Curr. Top. Dev. Biol. 2018, 127, 165–192.
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell. Biol. 2013, 14, 83–97.
- Jamart, C.; Naslain, D.; Gilson, H.; Francaux, M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am. J. Physiol.-Endocrinol. Metab. 2013, 305, 964–974.
- Louis, E.; Raue, U.; Yang, Y.; Jemiolo, B.; Trappe, S. Time course of proteolytic, cytokine, and myostatin gene expression after acute exercise in human skeletal muscle. J. Appl. Physiol. 2007, 103, 1744–1751.
- Pasiakos, S.M.; McClung, H.L.; McClung, J.P.; Urso, M.L.; Pikosky, M.A.; Cloutier, G.J.; Fielding, R.A.; Young, A.J. Molecular responses to moderate endurance exercise in skeletal muscle. Int. J. Sport Nutr. Exerc. Metab. 2010, 20, 282–290.
- Sanchez, A.M.J.; Candau, R.B.; Bernardi, H. FoxO transcription factors: Their roles in the maintenance of skeletal muscle homeostasis. Cell. Mol. Life Sci. 2014, 71, 1657–1671.
- Labeit, S.; Kohl, C.H.; Witt, C.C.; Labeit, D.; Jung, J.; Granzier, H. Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1-KO mice. J. Biomed. Biotechnol. 2010, 693741.
- Baehr, L.M.; Furlow, J.D.; Bodine, S.C. Muscle sparing in muscle RING finger 1 null mice: Response to synthetic glucocorticoids. J. Physiol. 2011, 589, 4759–4776.
- Koyama, S.; Hata, S.; Witt, C.C.; Ono, Y.; Lerche, S.; Ojima, K.; Chiba, T.; Doi, N.; Kitamura, F.; Tanaka, K.; et al. Muscle RING-Finger Protein-1 (MuRF1) as a Connector of Muscle Energy Metabolism and Protein Synthesis. J. Mol. Biol. 2008, 376, 1224–1236.
- Files, D.C.; D’Alessio, F.R.; Johnston, L.F.; Kesari, P.; Aggarwal, N.R.; Garibaldi, B.T.; Mock, J.R.; Simmers, J.L.; DeGorordo, A.; Murdoch, J.; et al. A Critical Role for Muscle Ring Finger-1 in Acute Lung Injury–associated Skeletal Muscle Wasting. Am. J. Respir. Crit. Care Med. 2012, 185, 825–834.
- Fielitz, J.; Kim, M.-S.; Shelton, J.M.; Latif, S.; Spencer, J.A.; Glass, D.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, R.N. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Investig. 2007, 117, 2486–2495.
- Polge, C.; Heng, A.; Jarzaguet, M.; Ventadour, S.; Claustre, A.; Combaret, L.; Béchet, D.; Matondo, M.; Uttenweiler-Joseph, S.; Monsarrat, B.; et al. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J. 2011, 25, 3790–3802.
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.-H.; Rockman, H.A.; Patterson, C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140.
- Polge, C.; Cabantous, S.; Deval, C.; Claustre, A.; Hauvette, A.; Bouchenot, C.; Aniort, J.; Béchet, D.; Combaret, L.; Attaix, D.; et al. A muscle-specific MuRF1-E2 network requires stabilization of MuRF1-E2 complexes by telethonin, a newly identified substrate. J. Cachexia Sarcopenia Muscle 2018, 9, 129–145.
- Rudolf, R.; Bogomolovas, J.; Strack, S.; Choi, K.R.; Khan, M.M.; Wagner, A.; Brohm, K.; Hanashima, A.; Gasch, A.; Labeit, D.; et al. Regulation of nicotinic acetylcholine receptor turnover by MuRF1 connects muscle activity to endo/lysosomal and atrophy pathways. Age 2013, 35, 1663–1674.
- Khan, M.M.; Strack, S.; Wild, F.; Hanashima, A.; Gasch, A.; Brohm, K.; Reischl, M.; Carnio, S.; Labeit, D.; Sandri, M.; et al. Role of autophagy, SQSTM1, SH3GLB1, and TRIM63 in the turnover of nicotinic acetylcholine receptors. Autophagy 2014, 10, 123–136.
- Li, H.-H.; Du, J.; Fan, Y.-N.; Zhang, M.-L.; Liu, D.-P.; Li, L.; Lockyer, P.; Kang, E.Y.; Patterson, C.; Willis, M.S. The Ubiquitin Ligase MuRF1 Protects Against Cardiac Ischemia/Reperfusion Injury by Its Proteasome-Dependent Degradation of Phospho-c-Jun. Am. J. Pathol. 2011, 178, 1043–1058.
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol.-Endocrinol. Metab. 2014, 307, E469–E484.
- Csibi, A.; Leibovitch, M.P.; Cornille, K.; Tintignac, L.A.; Leibovitch, S.A. MAFbx/Atrogin-1 Controls the Activity of the Initiation Factor eIF3-f in Skeletal Muscle Atrophy by Targeting Multiple C-terminal Lysines. J. Biol. Chem. 2009, 284, 4413–4421.
- Jogo, M.; Shiraishi, S.; Tamura, T.A. Identification of MAFbx as a myogenin-engaged F-box protein in SCF ubiquitin ligase. FEBS Lett. 2009, 583, 2715–2719.
- Lagirand-Cantaloube, J.; Cornille, K.; Csibi, A.; Batonnet-Pinchon, S.; Leibovitch, M.P.; Leibovitch, S.A. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS ONE 2009, 4, e4973.
- Wardle, F.C. Master control: Transcriptional regulation of mammalian Myod. J. Muscle Res. Cell Motil. 2019, 40, 211–226.
- Lokireddy, S.; Wijesoma, I.W.; Sze, S.K.; McFarlane, C.; Kambadur, R.; Sharma, M. Identification of atrogin-1-targeted proteins during the myostatin-induced skeletal muscle wasting. Am. J. Physiol.-Cell Physiol. 2012, 303, C512–C529.
- Romanello, V.; Sandri, M. Mitochondrial quality control and muscle mass maintenance. Front. Physiol. 2016, 2, 422.
- Zhang, J.; Xie, J.J.; Zhou, S.J.; Chen, J.; Hu, Q.; Pu, J.X.; Lu, J.-L. Diosgenin inhibits the expression of nedd4 in prostate cancer cells. Am. J. Transl. Res. 2019, 11, 3461–3471.
- Leermakers, P.A.; Schols, A.M.W.J.; Kneppers, A.E.M.; Kelders, M.C.J.M.; de Theije, C.C.; Lainscak, M.; Gosker, H.R. Molecular signalling towards mitochondrial breakdown is enhanced in skeletal muscle of patients with chronic obstructive pulmonary disease (COPD). Sci. Rep. 2018, 8, 1–13.
- Deval, C.; Calonne, J.; Coudy-Gandilhon, C.; Vazeille, E.; Bechet, D.; Polge, C.; Taillandier, D.; Attaix, D.; Combaret, L. Mitophagy and Mitochondria Biogenesis Are Differentially Induced in Rat Skeletal Muscles during Immobilization and/or Remobilization. Int. J. Mol. Sci. 2020, 21, 3691.
- Kang, C.; Yeo, D.; Ji, L.L. Muscle immobilization activates mitophagy and disrupts mitochondrial dynamics in mice. Acta Physiol. 2016, 218, 188–197.
- Balan, E.; Schwalm, C.; Naslain, D.; Nielens, H.; Francaux, M.; Deldicque, L. Regular Endurance Exercise Promotes Fission, Mitophagy, and Oxidative Phosphorylation in Human Skeletal Muscle Independently of Age. Front. Physiol. 2019, 10, 1088.
- Ehrlicher, S.E.; Stierwalt, H.D.; Miller, B.F.; Newsom, S.A.; Robinson, M.M. Mitochondrial adaptations to exercise do not require Bcl2-mediated autophagy but occur with BNIP3/Parkin activation. FASEB J. 2020, 34, 4602–4618.
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; LaStayo, P.C.; Marcus, R.L. Downregulation of E3 Ubiquitin Ligases and Mitophagy-Related Genes in Skeletal Muscle of Physically Inactive, Frail Older Women: A Cross-Sectional Comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048.
- Russ, D.W.; Wills, A.M.; Boyd, I.M.; Krause JWeakness, S.R. function and stress in gastrocnemius muscles of aged male rats. Exp. Gerontol. 2014, 50, 40–44.
- Marzetti, E.; Lorenzi, M.; Landi, F.; Picca, A.; Rosa, F.; Tanganelli, F.; Galli, M.; Doglietto, G.B.; Pacelli, F.; Cesari, M.; et al. Altered mitochondrial quality control signaling in muscle of old gastric cancer patients with cachexia. Exp. Gerontol. 2017, 87, 92–99.
- Chen, C.C.W.; Erlich, A.T.; Crilly, M.J.; Hood, D.A. Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E404–E415.
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function: Parkin is essential for optimal muscle and mitochondrial functions. J. Physiol. 2018, 596, 2565–2579.
- Ramesh, M.; Campos, J.C.; Lee, P.; Song, Y.; Hernandez, G.; Sin, J.; Tucker, K.C.; Saadaeijahromi, H.; Gurney, M.; Ferreira, J.C.B.; et al. Mitophagy protects against statin-mediated skeletal muscle toxicity. FASEB J. 2019, 33, 11857–11869.
- Furuya, N.; Ikeda, S.-I.; Sato, S.; Soma, S.; Ezaki, J.; Trejo, J.A.O.; Takeda-Ezaki, M.; Fujimura, T.; Arikawa-Hirasawa, E.; Tada, N.; et al. PARK2/Parkin-mediated mitochondrial clearance contributes to proteasome activation during slow-twitch muscle atrophy via NFE2L1 nuclear translocation. Autophagy 2014, 10, 631–641.
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083.
- Yu, R.; Chen, J.A.; Xu, J.; Cao, J.; Wang, Y.; Thomas, S.S.; Hu, Z. Suppression of muscle wasting by the plant-derived compound ursolic acid in a model of chronic kidney disease. J. Cachexia Sarcopenia Muscle 2017, 8, 327–341.
- Hahn, A.; Kny, M.; Pablo-Tortola, C.; Todiras, M.; Willenbrock, M.; Schmidt, S.; Schmoeckel, K.; Jorde, I.; Nowak, M.; Jarosch, E.; et al. Serum amyloid A1 mediates myotube atrophy via Toll-like receptors. J. Cachexia Sarcopenia Muscle 2020, 11, 103–119.
- Yoo, S.-H.; Mohawk, J.A.; Siepka, S.M.; Shan, Y.; Huh, S.K.; Hong, H.-K.; Kornblum, I.; Kumar, V.; Koike, N.; Xu, M.; et al. Competing E3 Ubiquitin Ligases Govern Circadian Periodicity by Degradation of CRY in Nucleus and Cytoplasm. Cell 2013, 152, 1091–1105.
- Lucas, X.; Ciulli, A. Recognition of substrate degrons by E3 ubiquitin ligases and modulation by small-molecule mimicry strategies. Curr. Opin. Struct. Biol. 2017, 44, 101–110.
- Kwak, K.S.; Zhou, X.; Solomon, V.; Baracos, V.E.; Davis, J.; Bannon, A.W.; Boyle, W.J.; Lacey, D.L.; Han, H.Q. Regulation of protein catabolism by muscle-specific and cytokine-inducible ubiquitin ligase E3alpha-II during cancer cachexia. Cancer Res. 2004, 64, 8193–8198.
- Seaborne, R.A.; Hughes, D.C.; Turner, D.C.; Owens, D.J.; Baehr, L.M.; Gorski, P.; Semenova, E.A.; Borisov, O.V.; Larin, A.K.; Popov, D.V.; et al. UBR5 is a novel E3 ubiquitin ligase involved in skeletal muscle hypertrophy and recovery from atrophy. J. Physiol. 2019, 597, 3727–3749.
- Besche, H.C.; Haas, W.; Gygi, S.P.; Goldberg, A.L. Isolation of Mammalian 26S Proteasomes and p97/VCP Complexes Using the Ubiquitin-like Domain from HHR23B Reveals Novel Proteasome-Associated Proteins. Biochemistry 2009, 48, 2538–2549.
- Morén, A.; Imamura, T.; Miyazono, K.; Heldin, C.H.; Moustakas, A. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J. Biol. Chem. 2005, 280, 22115–22123.
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480.
- Tényi, Á.; Cano, I.; Marabita, F.; Kiani, N.; Kalko, S.G.; Barreiro, E.; de Atauri, P.; Cascante, M.; Gomez-Cabrero, D.; Roca, J. Network modules uncover mechanisms of skeletal muscle dysfunction in COPD patients. J. Transl. Med. 2018, 16, 1–12.
- Xin, H.; Xu, X.; Li, L.; Ning, H.; Rong, Y.; Shang, Y.; Wang, Y.; Fu, X.-Y.; Chang, Z. CHIP controls the sensitivity of transforming growth factor-β signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J. Biol. Chem. 2005, 280, 20842–20850.
- Li, R.F.; Shang, Y.; Liu, D.; Ren, Z.S.; Chang, Z.; Sui, S.F. Differential Ubiquitination of Smad1 Mediated by CHIP: Implications in the Regulation of the Bone Morphogenetic Protein Signaling Pathway. J. Mol. Biol. 2007, 374, 777–790.
- Schisler, J.C.; Patterson, C.; Willis, M.S. Skeletal Muscle Mitochondrial Alterations in Carboxyl Terminus of HSC70 Interacting Protein (CHIP)−/− Mice. Afr. J. Cell. Pathol. 2016, 6, 28–36.
- Chen, N.; Balasenthil, S.; Reuther, J.; Frayna, A.; Wang, Y.; Chandler, D.S.; Abruzzo, L.V.; Rashid, A.; Rodriguez, J.; Lozano, G.; et al. DEAR1 is a chromosome 1p35 tumor suppressor and master regulator of TGF-β-driven epithelial-mesenchymal transition. Cancer Discov. 2013, 3, 1172–1189.
- Schmidt, F.; Kny, M.; Zhu, X.; Wollersheim, T.; Persicke, K.; Langhans, C.; Lodka, D.; Kleber, C.; Weber-Carstens, S.; Fielitz, J. The E3 ubiquitin ligase TRIM62 and inflammationinduced skeletal muscle atrophy. Crit. Care 2014, 18, 1–12.
- He, B.; Tang, R.H.; Weisleder, N.; Xiao, B.; Yuan, Z.; Cai, C.; Zhu, H.; Lin, P.; Qiao, C.; Li, J.; et al. Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure inδ-sarcoglycan-deficient hamsters. Mol. Ther. 2012, 20, 727–735.
- Gushchina, L.V.; Bhattacharya, S.; McElhanon, K.E.; Choi, J.H.; Manring, H.; Beck, E.X.; Alloush, J.; Weisleder, N. Treatment with Recombinant Human MG53 Protein Increases Membrane Integrity in a Mouse Model of Limb Girdle Muscular Dystrophy 2B. Mol. Ther. 2017, 25, 2360–2371.
- Cao, C.M.; Zhang, Y.; Weisleder, N.; Ferrante, C.; Wang, X.; Lv, F.; Zhang, Y.; Song, R.; Hwang, M.; Jin, L.; et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation 2010, 121, 2565–2574.



