 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rawan Altalhi | + 1939 word(s) | 1939 | 2021-05-26 11:26:12 | | | |
| 2 | Nora Tang | Meta information modification | 1939 | 2021-05-31 03:02:03 | | |
Video Upload Options
Hypofibrinolysis is a key abnormality in diabetes and contributes to the adverse vascular outcome in this population. Plasminogen activator inhibitor (PAI)-1 is an important regulator of the fibrinolytic process and levels of this antifibrinolytic protein are elevated in diabetes and insulin resistant states. This review describes both the physiological and pathological role of PAI-1 in health and disease, focusing on the mechanism of action as well as protein abnormalities in vascular disease with special focus on diabetes. Attempts at inhibiting protein function, using different techniques, are also discussed including direct and indirect interference with production as well as inhibition of protein function. Developing PAI-1 inhibitors represents an alternative approach to managing hypofibrinolysis by targeting the pathological abnormality rather than current practice that relies on profound inhibition of the cellular and/or acellular arms of coagulation, and which can be associated with increased bleeding events. The review offers up-to-date knowledge on the mechanisms of action of PAI-1 together with the role of altering protein function to improve hypofirbinolysis. Developing PAI-1 inhibitors may form for the basis of future new class of antithrombotic agents that reduce vascular complications in diabetes.
1. Indirect Modulation of PAI-1 Production or Function
1.1. A. Non-Pharmacological Intervention to Lower PAI-1 Levels
Weight Loss (Healthy Diet and Physical Exercise)
Weight loss is one of the best methods to improve obesity-related fibrinolytic impairment [1], partly related to a marked decrease in PAI-1 production, which has been demonstrated in a number of studies [2]. It has also been shown that not only weight loss but also increased physical activity are linked to decreased PAI-1 levels [3]. Importantly, the decrease in PAI-1 levels is associated with improved clinical parameters; Killewich et al. [4] found that patients with intermittent claudication, due to peripheral artery disease, had improvement in their symptoms after a programme of physical exercise, which was associated with decreased PAI-1 activity and enhanced fibrinolytic activity. Interestingly, those with higher baseline PAI-1 levels gained the most clinical benefit from the intervention, directly implicating protein levels in disease presentation [5].
1.2. Pharmacological Interventions to Lower PAI-1 Levels
Renin-Angiotensin System Blockers
The activation of the renin–angiotensin system is closely related to PAI-1 [6]. Angiotensin II (also produced by adipose tissue) is a biologically active angiotensinogen processing agent and has been shown to stimulate PAI-1 expression at the transcriptional level in human adipocytes [7]. The effects of angiotensin-converting enzyme (ACE) inhibitors on cardio protection appear to be mediated by pathways that are partially independent of the reduction in blood pressure. Vaughan et al. reported that one proposed mechanism is related to modulation of PAI-1 concentrations [8]. However, others failed to show an effect of ACE inhibitors on PAI-1 levels [9]. Therefore, this remains an unresolved area and future work is required to understand the role of ACE inhibitors on fibrinolysis in general and PAI-1 activity/levels in particular [10].
Statins
Anti-hyperlipidaemic agents, in particular statins, have been shown to modulate the prothrombotic environment and affect the fibrinolytic system [11]. Statins inhibit the expression of PAI-1 and their use is associated with reduction in PAI-1 levels, which may explain improved t-PA activity in those treated with these agents [12][13][14].
Fibrates
Fibrates can decrease the in vitro production of PAI-1 independently of the triglyceride reducing effect [15]. In vivo use of fibrates also decreases PAI-1 levels and reduces protein expression in human arterial smooth muscle cells (SMC) [15][16]. Different fibrates showed different potencies to suppress PAI-1 synthesis, but these effects are generally mild and the clinical role of these observations remain unclear [17].
Insulin-Sensitising Agents
Insulin-sensitising agents, such as metformin and thiazolidinediones have been shown to have beneficial effects on the fibrinolytic system, mediated, at least in part, by a reduction in PAI-1 levels [18]. Glucagon-like peptide-1 receptor agonists inhibit TNF-α stimulated PAI-1 production in vitro [19], while metformin reduces PAI-1 levels in vivo, an effect believed to be related to enhanced insulin sensitivity and/or reduction in proinsulin production [20][21][22].
2. Direct Modulation of PAI-1 Production or Activity
Many research groups have invested in the development of PAI-1 inhibitors over the past three decades [23] either through inhibiting PAI-1 synthesis or interfering with serpin-proteinase interaction (Figure 5). Despite large volume of research in this area, no PAI-1 inhibitor is yet available for routine clinical use.
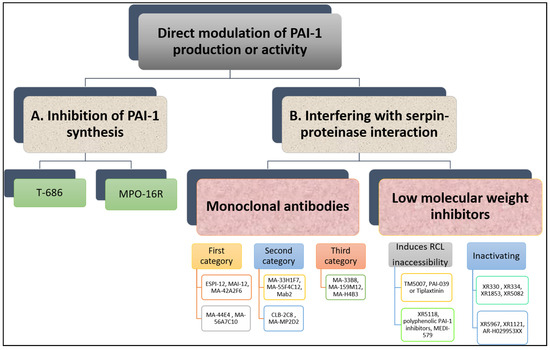
Figure 5. Overview of direct modulation of PAI-1 production or activity by (A) inhibition of PAI-1 synthesis or (B) interfering with serpin–proteinase interaction.
2.1. Inhibition of PAI-1 Synthesis
Reduction in PAI-1 synthesis represents an attractive approach to enhancing fibrinolysis and reducing thrombosis risk. However, this needs to be undertaken in a controlled manner as full inhibition of PAI-1 synthesis predisposes to mild-severe bleeding events, supported by evidence from individuals with complete PAI-1 deficiency [24]. Therefore, ideally agents suppressing PAI-1 production should reduce excess PAI-1 levels to attain normal levels and restore normal haemostasis [25].
In vitro studies have shown that TGF-β-stimulated PAI-1 production in cultured endothelial cells is attenuated by a butadiene derivative, T-686. Furthermore, T-686 also diminished the rise in PAI-1 activity in aortic atherosclerotic lesions of rabbits. This was accompanied by decreased aortic PAI-1 mRNA expression, associated with a reduction in the development of atherosclerotic lesions [26]. Pawlowska et al. [27] showed that an oligonucleotide antisense to PAI-1 mRNA (MPO-16R) decreased PAI-1 concentration in rat plasma and platelets, while MPO-16R induced a large delay in occlusion time in an experimental model of rat arterial thrombosis. Therefore, reducing PAI-1 production is possible and preliminary evidence suggests this strategy can alter the atherothrombotic process. However, findings from these studies have not been translated into work in man and the clinical future of this antithrombotic strategy is uncertain.
2.2. Interfering with Serpin-Proteinase Interaction
Although the available evidence is very preliminary, studies have shown that inhibition of PAI-1 enhances endogenous fibrinolytic activity without directly affecting blood coagulation or platelet function [28]. Several PAI-1 inhibitors have been studied in vitro and in vivo and have shown variable antithrombotic efficacy.
Monoclonal Antibodies
A variety of monoclonal antibodies against human PAI-1 has been developed to modulate protein activity [29]. These can be subdivided into at least three different categories. First, monoclonal antibodies may prevent the formation of the initial Michaelis complex formed between PAI-1 and its target protease. It was found that monoclonal antibodies against PAI-1, including ESPI-12, MAI-12, MA-42A2F6, MA-44E4 and MA-56A7C10, with their epitopes located near the RCL of PAI-1, interfere directly with the PAI-1/protease interaction [30]. MAI-12, one of the PAI-1 binding antibodies that inhibit complex formation with the target, has been studied extensively in vitro and in an in vivo venous thrombosis model [31]. MAI-12 inhibited human and rabbit PAI-1 in vitro and significantly enhanced endogenous thrombolysis in a rabbit model of jugular vein thrombosis and partially prevented thrombus extension [31].
A second category of PAI-1 inhibiting antibodies are the so-called “switching antibodies”, which inhibit PAI-1 activity by cleaving and inactivating the protein. These antibodies include MA-33H1F7, MA-55F4C12 [32], Mab2 [33], CLB-2C8 [34] and MA-MP2D2 [35]. The murine monoclonal antihuman PAI-1 antibody CLB-2C8 showed great promise as it inhibited protein activity in different species [36], while in vivo administration significantly enhanced endogenous thrombolysis, and reduced thrombus growth in a rabbit jugular vein thrombosis model [36]. Despite the early promise, CLB-2C8 was not followed up with human studies for reasons that are unclear.
As for the third category of PAI-1 inhibiting antibodies, these are known for accelerating conversion of active PAI-1 to its latent conformation and include MA-H4B3, MA-159M12 and MA-33B8 antibodies [37]. The monoclonal antibody, MA-33B8, was reported to convert PAI-1 to latent conformation and random mutagenesis was used to identify the MA-33B8 epitope in the PAI-1 molecule to comprehend the mechanism of action [38]. Surface plasmon resonance showed 100-fold higher affinity of MA-33B8 to latent PAI-1 than active protein [38]. Additionally, structural modelling results specified the presence of a particular intermediate structure of PAI-1 that is stabilised by MA-33B8 binding [38]. It was suggested that this intermediate form of PAI-1 has a partial insertion into β-sheet A of the reactive centre loop, supporting the hypothesis that active serpins have flexible RCLs, and that this flexibility is crucial for the role of the inhibitor [39]. As a result, the activity of the serpin may also be blocked by synthetic peptides homologous to the RCL serpin converting PAI-1 to a shape similar to the latent form [40]. Moreover, Ngo et al. [41] reported that antibody MA-159M12 promoted the conversion of active to latent conformation, further indicating this is a viable approach to alter PAI-1 activity. Antibody MA-H4B3 has been shown to inactivate recombinant PAI-1 in a time-dependent way [42], which indicates that, in the presence of MA-H4B3, incorporation of the RCL is accelerated, suggesting that the loss of activity is the result of the transition of latency [42].
Low Molecular Weight Inhibitors
Several PAI-1 low molecular weight antagonists have been discovered and characterised. The first were diketopiperazines (XR330 and XR334) and, from that template, more potent antagonists were designed (XR1853, XR5082, XR5967, XR1121 and XR5118) which inhibit PAI-1 by inducing the transition from active PAI-1 to non-reactive PAI-1 [43][44]. XR334, XR1853 and XR5082 effectively increased fibrinolysis in vivo by inhibiting the interaction of t-PA/u-PA and PAI-1 in a rat carotid artery thrombosis model [45].
XR5118 administration improved ex vivo fibrinolysis and protected against thrombus formation in a rat electrically stimulated carotid artery (ESCA) model [46][47]. The protection in this arterial thrombosis was associated with a significant decrease in PAI-1 activity and a significant increase in t-PA activity in plasma [46][47]. Importantly, inhibition of PAI-1 by XR5118 did not contribute to any increase in rat bleeding time, which held promise for the safety of this compound [47].
Furthermore, TM5007, a derivative of indole oxoacetic acid was shown to effectively inhibit PAI-1 activity [28]. This compound is metabolically stable, non-toxic, and has demonstrated strong oral bioavailability and in vivo efficacy in a rat thrombosis model [28].
Björquist et al. [48] developed the flufenamic acid-based AR-H029953XX after it was reported that flufenamic acid and its derivatives enhanced in vitro plasma clot lysis in an assay of plasmin generation [49].
As for PAI-039 or Tiplaxtinin, an indole derivative, seemed a very promising PAI-1 inhibitor [50]. It has been examined in several animal models confirming a putative value of PAI-1 inhibition for therapeutic purposes [51]. In another study, however, Tiplaxtinin did not inhibit the activity of PAI-1 in rat plasma 15 min after intravenous injection, which was suggested to be due to the fact that Tiplaxtinin is unable to inhibit vitronectin bound PAI-1 because of the overlapping epitopes of vitronectin and Tiplaxtinin on PAI-1 [52].
A novel class of polyphenolic PAI-1 inhibitors was identified by Cale et al. [53], representing an enhanced 10–1000-fold potency over previously described PAI-1 inhibitors. This class exerted its PAI-1-inhibitor effect by blocking the initial bond between the protein and protease. These compounds inactivate PAI-1 in the presence of vitronectin, giving them an advantage over other PAI-1 inhibitors. Two of these compounds demonstrated efficacy in plasma and one other blocked PAI-1 activity in in vivo mice studies. The authors proposed that the known cardiovascular advantages of dietary polyphenols may be due in part to PAI-1 inactivation, although this is only a hypothesis that is yet to be tested. A human antibody, MEDI-579, has recently been shown to bind to the active form of human PAI-1 with high affinity and specificity [54]. Importantly, MEDI-579 effects were still evident in vitronectin bound PAI-1. Crystallography demonstrated that this specificity is achieved due to the attachment of MEDI-579 Fab to the RCL of PAI-1 and to the same exosite used by both t-PA and u-PA [54]. This indicates that MEDI-579 is able to modulate the interaction of PAI-1 with t-PA and u-PA in a manner not previously defined for a human PAI-1 inhibitor by directly competing with proteases for RCL binding.
References
- Skurk, T.; Hauner, H. Obesity and impaired fibrinolysis: Role of adipose production of plasminogen activator inhibitor-1. Int. J. Obes. 2004, 28, 1357–1364.
- McGill, H.C., Jr.; McMahan, C.A.; Herderick, E.E.; Zieske, A.W.; Malcom, G.T.; Tracy, R.E.; Strong, J.P. Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation 2002, 105, 2712–2718.
- Cesari, M.; Pahor, M.; Incalzi, R.A. REVIEW: Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions. Cardiovasc. Ther. 2010, 28, e72–e91.
- Killewich, L.A.; Macko, R.F.; Montgomery, P.S.; Wiley, L.A.; Gardner, A.W. Exercise training enhances endogenous fibrinolysis in peripheral arterial disease. J. Vasc. Surg. 2004, 40, 741–745.
- Kokkinos, P.; Myers, J. Exercise and physical activity: Clinical outcomes and applications. Circulation 2010, 122, 1637–1648.
- Brown, N.J.; Agirbasli, M.A.; Williams, G.H.; Litchfield, W.R.; Vaughan, D.E. Effect of Activation and Inhibition of the Renin-Angiotensin System on Plasma PAI-1. Hypertension 1998, 32, 965–971.
- Skurk, T.; Lee, Y.-M.; Hauner, H. Angiotensin II and Its Metabolites Stimulate PAI-1 Protein Release From Human Adipocytes in Primary Culture. Hypertension 2001, 37, 1336–1340.
- Vaughan, D.E.; Rouleau, J.-L.; Ridker, P.M.; Arnold, J.M.O.; Menapace, F.J.; Pfeffer, M.A. Effects of Ramipril on Plasma Fibrinolytic Balance in Patients With Acute Anterior Myocardial Infarction. Circulation 1997, 96, 442–447.
- Cesari, M.; Kritchevsky, S.B.; Atkinson, H.H.; Penninx, B.W.; Di Bari, M.; Tracy, R.P.; Pahor, M. Angiotensin-converting enzyme inhibition and novel cardiovascular risk biomarkers: Results from the Trial of Angiotensin Converting Enzyme Inhibition and Novel Cardiovascular Risk Factors (TRAIN) study. Am. Heart J. 2009, 157, 334.e1–334.e8.
- Skurk, T.; Lee, Y.-M.; Nicuta-Rolfs, T.-O.; Haastert, B.; Wirth, A.; Hauner, H. Effect of the angiotensin II receptor blocker candesartan on fibrinolysis in patients with mild hypertension. Diabetes Obes. Metab. 2004, 6, 56–62.
- Zirlik, A.; Ernst, S.; Leugers, A.; Willecke, F.; Sobel, B.E.; Bode, C.; Nordt, T.K. Inhibition by fibrates of plasminogen activator inhibitor type-1 expression in human adipocytes and preadipocytes. Thromb. Haemost. 2009, 101, 1060–1069.
- Simone, T.M.; Higgins, P.J. Low Molecular Weight Antagonists of Plasminogen Activator Inhibitor-1: Therapeutic Potential in Cardiovascular Disease. Mol. Med. Ther. 2012, 1, 101.
- Lindahl, T.L.; Sigurdardottir, O.; Wiman, B. Stability of plasminogen activator inhibitor 1 (PAI-1). Thromb. Haemost. 1989, 61, 748–751.
- Gils, A.; Declerck, P.J. Proteinase Specificity and Functional Diversity in Point Mutants of Plasminogen Activator Inhibitor 1. J. Biol. Chem. 1997, 272, 12662–12666.
- Baluta, M.M.; Vintila, M.M. PAI-1 inhibition—Another therapeutic option for cardiovascular protection. Maedica 2015, 10, 147.
- Oishi, K.; Koyanagi, S.; Matsunaga, N.; Kadota, K.; Ikeda, E.; Hayashida, S.; Kuramoto, Y.; Shimeno, H.; Soeda, S.; Ohdo, S. Bezafibrate Induces Plasminogen Activator Inhibitor-1 Gene Expression in a CLOCK-Dependent Circadian Manner. Mol. Pharmacol. 2010, 78, 135–141.
- Erickson, L.A.; Ginsberg, M.H.; Loskutoff, D.J. Detection and partial characterization of an inhibitor of plasminogen activator in human platelets. J. Clin. Investig. 1984, 74, 1465–1472.
- Kruszynska, Y.T.; Yu, J.G.; Olefsky, J.M.; Sobel, B.E. Effects of troglitazone on blood concentrations of plasminogen activator inhibitor 1 in patients with type 2 diabetes and in lean and obese normal subjects. Diabetes 2000, 49, 633–639.
- Liu, H.; E Dear, A.; Knudsen, L.B.; Simpson, R.W. A long-acting glucagon-like peptide-1 analogue attenuates induction of plasminogen activator inhibitor type-1 and vascular adhesion molecules. J. Endocrinol. 2009, 201, 59–66.
- Schneider, D.J. Abnormalities of coagulation, platelet function, and fibrinolysis associated with syndromes of insulin resistance. Coron. Artery Dis. 2005, 16, 473–476.
- Grant, P.J. The effects of metformin on the fibrinolytic system in diabetic and non-diabetic subjects. Diabete Metab. 1991, 17, 168–173.
- Nagi, D.; Ali, V.M.; Yudkin, J. Effect of Metformin on Intact Proinsulin and des 31,32 Proinsulin Concentrations in Subjects with Non-insulin-dependent (Type 2) Diabetes Mellitus. Diabet. Med. 1996, 13, 753–757.
- Fortenberry, Y.M. Plasminogen activator inhibitor-1 inhibitors: A patent review (2006—Present). Expert Opin. Ther. Pat. 2013, 23, 801–815.
- Khan, S.S.; Shah, S.J.; Klyachko, E.; Baldridge, A.S.; Eren, M.; Place, A.T.; Aviv, A.; Puterman, E.; Lloyd-Jones, D.M.; Heiman, M.; et al. A null mutation inSERPINE1protects against biological aging in humans. Sci. Adv. 2017, 3, eaao1617.
- Vaughan, D.E. PAI-1 antagonists: The promise and the peril. Trans. Am. Clin. Climatol. Assoc. 2011, 122, 312.
- Vinogradsky, B.; Bell, S.P.; Woodcock-Mitchell, J.; Ohtani, A.; Fujii, S. A new butadiene derivative, T-686, inhibits plasminogen activator inhibitor type-1 production in vitro by cultured human vascular endothelial cells and development of atherosclerotic lesions in vivo in rabbits. Thromb. Res. 1997, 85, 305–314.
- Pawlowska, Z.; Chabielska, E.; Kobylańska, A.; Maciaszek, A.; Swiatkowska, M.; Buczko, W.; Stec, W.; Cierniewski, C. Regulation of PAI-1 Concentration in Platelets by Systemic Administration of Antisense Oligonucleotides to Rats. Thromb. Haemost. 2001, 85, 1086–1089.
- Izuhara, Y.; Takahashi, S.; Nangaku, M.; Takizawa, S.; Ishida, H.; Kurokawa, K.; van Ypersele de Strihou, C.; Hirayama, N.; Miyata, T. Inhibition of plasminogen activator inhibitor-1: Its mechanism and effectiveness on coagulation and fibrosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 672–677.
- Gils, A.; Declerck, P.J. The structural basis for the pathophysiological relevance of PAI-1 in cardiovascular diseases and the development of potential PAI-1 inhibitors. Thromb. Haemost. 2004, 91, 425–437.
- Bijnens, A.P.; Gils, A.; Stassen, J.M.; Komissarov, A.A.; Knockaert, I.; Brouwers, E.; Shore, J.D.; Declerck, P.J. The Distal Hinge of the Reactive Site Loop and Its Proximity a Target to Modulate Plasminogen Activator Inhibitor-1 Activity. J. Biol. Chem. 2001, 276, 44912–44918.
- Levi, M.; Biemond, B.J.; van Zonneveld, A.J.; Cate, J.W.T.; Pannekoek, H. Inhibition of plasminogen activator inhibitor-1 activity results in promotion of endogenous thrombolysis and inhibition of thrombus extension in models of experimental thrombosis. Circulation 1992, 85, 305–312.
- Bijnens, A.-P.; Gils, A.; Knockaert, I.; Stassen, J.M.; Declerck, P.J. Importance of the Hinge Region between α-Helix F and the Main Part of Serpins, Based upon Identification of the Epitope of Plasminogen Activator Inhibitor Type 1 Neutralizing Antibodies. J. Biol. Chem. 2000, 275, 6375–6380.
- Wind, T.; Jensen, M.A.; Andreasen, P.A. Epitope mapping for four monoclonal antibodies against human plasminogen activator inhibitor type-1: Implications for antibody-mediated PAI-1-neutralization and vitronectin-binding. Eur. J. Biochem. 2001, 268, 1095–1106.
- Stoop, A.; Jespers, L.; Lasters, I.; Eldering, E.; Pannekoek, H. High-density mutagenesis by combined DNA shuffling and phage display to assign essential amino acid residues in protein-protein interactions: Application to study structure-function of plasminogen activation inhibitor 1 (PAI-I). J. Mol. Biol. 2000, 301, 1135–1147.
- van de Craen, B.; Scroyen, I.; Abdelnabi, R.; Brouwers, E.; Lijnen, H.R.; Declerck, P.J.; Gils, A. Characterization of a panel of monoclonal antibodies toward mouse PAI-1 that exert a significant profibrinolytic effect in vivo. Thromb. Res. 2011, 128, 68–76.
- Biemond, B.J.; Levi, M.; Coronel, R.; Janse, M.J.; Ten Cate, J.W.; Pannekoek, H. Thrombolysis and reocclusion in experimental jugular vein and coronary artery thrombosis: Effects of a plasminogen activator inhibitor type 1-neutralizing monoclonal antibody. Circulation 1995, 91, 1175–1181.
- Naessens, D.; Gils, A.; Compernolle, G.; Declerck, P. Elucidation of the epitope of a latency-inducing antibody: Identification of a new molecular target for PAI-1 inhibition. Thromb. Haemost. 2003, 90, 52–58.
- Gorlatova, N.V.; Elokdah, H.; Fan, K.; Crandall, D.L.; Lawrence, D.A. Mapping of a conformational epitope on plasminogen activator inhibitor-1 by random mutagenesis: Implications for serpin function. J. Biol. Chem. 2003, 278, 16329–16335.
- Lawrence, D.A.; Olson, S.T.; Muhammad, S.; Day, D.E.; Kvassman, J.-O.; Ginsburg, D.; Shore, J.D. Partitioning of Serpin-Proteinase Reactions between Stable Inhibition and Substrate Cleavage Is Regulated by the Rate of Serpin Reactive Center Loop Insertion into β-Sheet A. J. Biol. Chem. 2000, 275, 5839–5844.
- Kvassman, J.-O.; Lawrence, D.A.; Shore, J.D. The Acid Stabilization of Plasminogen Activator Inhibitor-1 Depends on Protonation of a Single Group That Affects Loop Insertion into β-Sheet A. J. Biol. Chem. 1995, 270, 27942–27947.
- Ngo, T.-H.; Zhou, Y.; Stassen, J.-M.; Declerck, P. Importance of N-Terminal Residues in Plasminogen Activator Inhibitor 1 on its Antibody Induced Latency Transition. Thromb. Haemost. 2002, 88, 288–293.
- Dupont, D.M.; Blouse, G.E.; Hansen, M.; Mathiasen, L.; Kjelgaard, S.; Jensen, J.K.; Christensen, A.; Gils, A.; Declerck, P.J.; Andreasen, P.A.; et al. Evidence for a Pre-latent Form of the Serpin Plasminogen Activator Inhibitor-1 with a Detached β-Strand 1C. J. Biol. Chem. 2006, 281, 36071–36081.
- Folkes, A.; Roe, M.B.; Sohal, S.; Golec, J.; Faint, R.; Brooks, T.; Charlton, P. Synthesis and in vitro evaluation of a series of diketopiperazine inhibitors of plasminogen activator inhibitor-1. Bioorg. Med. Chem. Lett. 2001, 11, 2589–2592.
- Bryans, J.; Charlton, P.; Chicarelli-Robinson, I.; Collins, M.; Faint, R.; Latham, C.; Shaw, I.; Trew, S. Inhibition of Plasminogen Activator Inhibitor-1 Activity by Two Diketopiperazines, XR330 and XR334 Produced by Streptomyces sp. J. Antibiot. 1996, 49, 1014–1021.
- Charlton, P.A.; Faint, R.W.; Bent, F.; Bryans, J.; Chicarelli-Robinson, I.; Mackie, I.; Machin, S.; Bevan, P. Evaluation of a low molecular weight modulator of human plasminogen activator inhibitor-1 activity. Thromb. Haemost. 1996, 76, 808–815.
- Charlton, P. The status of plasminogen activator inhibitor-1 as a therapeutic target. Expert Opin. Investig. Drugs 1997, 6, 539–554.
- Friederich, P.W.; Levi, M.; Biemond, B.J.; Charlton, P.; Templeton, D.; van Zonneveld, A.J.; Bevan, P.; Pannekoek, H.; Cate, J.W.T. Novel low-molecular-weight inhibitor of PAI-1 (XR5118) promotes endogenous fibrinolysis and reduces postthrombolysis thrombus growth in rabbits. Circulation 1997, 96, 916–921.
- Björquist, P.; Ehnebom, J.; Inghardt, T.; Hansson, L.; Lindberg, M.; Linschoten, M.; Strömqvist, M.; Deinum, J. Identification of the Binding Site for a Low-Molecular-Weight Inhibitor of Plasminogen Activator Inhibitor Type 1 by Site-Directed Mutagenesis. Biochemistry 1998, 37, 1227–1234.
- Gils, A.; Stassen, J.-M.; Nar, H.; Kley, J.; Wienen, W.; Ries, U.; Declerck, P. Characterization and Comparative Evaluation of a Novel PAI-1 Inhibitor. Thromb. Haemost. 2002, 88, 137–143.
- Elokdah, H.; Abou-Gharbia, M.; Hennan, J.K.; McFarlane, G.; Mugford, C.P.; Krishnamurthy, G.; Crandall, D.L. Tiplaxtinin, a Novel, Orally Efficacious Inhibitor of Plasminogen Activator Inhibitor-1: Design, Synthesis, and Preclinical Characterization. J. Med. Chem. 2004, 47, 3491–3494.
- Lee, S.H.; Eren, M.; Vaughan, U.E.; Schleimer, R.P.; Cho, S.H. A Plasminogen Activator Inhibitor-1 Inhibitor Reduces Airway Remodeling in a Murine Model of Chronic Asthma. Am. J. Respir. Cell Mol. Biol. 2012, 46, 842–846.
- Rupin, A.; Gaertner, R.; Mennecier, P.; Richard, I.; Benoist, A.; de Nanteuil, G.; Verbeuren, T.J. S35225 is a direct inhibitor of Plasminogen Activator Inhibitor type-1 activity in the blood. Thromb. Res. 2008, 122, 265–270.
- Cale, J.M.; Li, S.-H.; Warnock, M.; Su, E.J.; North, P.R.; Sanders, K.L.; Puscau, M.M.; Emal, C.D.; Lawrence, D.A. Characterization of a Novel Class of Polyphenolic Inhibitors of Plasminogen Activator Inhibitor-1. J. Biol. Chem. 2010, 285, 7892–7902.
- Vousden, K.A.; Lundqvist, T.; Popovic, B.; Naiman, B.; Carruthers, A.M.; Newton, P.; Johnson, D.J.D.; Pomowski, A.; Wilkinson, T.; Dufner, P.; et al. Discovery and characterisation of an antibody that selectively modulates the inhibitory activity of plasminogen activator inhibitor-1. Sci. Rep. 2019, 9, 1–12.


