Hutchinson-Gilford progeria syndrome (HGPS) is a rare genetic disease that recapitulates many symptoms of physiological aging and precipitates death. Patients develop severe vascular alterations, mainly massive vascular smooth muscle cell loss, vessel stiffening, calcification, fibrosis, and generalized atherosclerosis, as well as electrical, structural, and functional anomalies in the heart. As a result, most HGPS patients die of myocardial infarction, heart failure, or stroke typically during the first or second decade of life. No cure exists for HGPS, and therefore it is of the utmost importance to define the mechanisms that control disease progression in order to develop new treatments to improve the life quality of patients and extend their lifespan. Since the discovery of the HGPS-causing mutation, several animal models have been generated to study multiple aspects of the syndrome and to analyze the contribution of different cell types to the acquisition of the HGPS-associated cardiovascular phenotype.
1. Hutchinson-Gilford Progeria Syndrome (HGPS)
HGPS (also termed progeria) is a rare genetic disorder (estimated prevalence of 1 in 18–20 million people) caused by a mutation in the
LMNA gene that provokes accelerated aging and premature death
[1][2][3]. In mammals,
LMNA normally encodes two major alternatively spliced A-type lamin variants, lamin A and lamin C, which are main components of the nuclear lamina. Post-translational modifications of prelamin A that yield mature lamin A include C-terminal farnesylation and carboxymethylation and subsequent cleavage of the modified C-terminus by the zinc metalloprotease ZMPSTE24. “Classical” progeria is caused by a heterozygous de novo c.1824C>T (p.G608G) point mutation in the
LMNA gene. This creates an aberrant splice site in exon 11 that deletes 150 nucleotides, resulting in the synthesis of a truncated prelamin A variant called progerin that lacks the cleavage site needed to remove the farnesylated and carboxymethylated C-terminus
[4]. Progerin is expressed in most differentiated cells and induces multiple cellular and physiological anomalies, including defects in nuclear morphology, chromatin disorganization, elevated DNA damage, impaired stem cell maintenance and differentiation, metabolic alterations, autophagy deregulation, and systemic inflammation
[5].
HGPS patients appear normal at birth, but start to develop a premature and accelerated aging phenotype between 1 and 2 years of age. Typical features include failure to thrive, alopecia, loss of subcutaneous fat, skin alterations, tooth and bone abnormalities, muscle weakness, and progressive joint contractures. However, the most clinically relevant feature of HGPS is the development of generalized atherosclerosis and cardiac dysfunction, which ultimately lead to death mainly due to myocardial infarction, heart failure, or stroke at an average age of 14.6 years
[4][6]. The following section describes the HGPS-associated cardiovascular phenotype in detail.
2. The Cardiovascular Phenotype in HGPS Patients: A Historical Perspective
Cardiovascular disease (CVD) is the main cause of death in HPGS, and it is therefore crucial to decipher the underlying mechanisms in order to design effective therapies. Because HGPS patients typically lack many of the classical cardiovascular risk factors such as hypercholesterolemia, high C-reactive protein, obesity, and smoking, the study of the mechanisms underlying CVD in HGPS also presents an opportunity to understand CVD in non-HGPS individuals in relative isolation from confounding risk factors. A robust characterization of the HGPS cardiovascular phenotype is also critical for developing clinical standards of care and objective cardiovascular readouts of therapeutic efficacy in HGPS clinical trials.
The extreme rarity of HGPS hinders systematic cardiovascular examination of patients, because the very few HGPS patients are spread around the world, may be diagnosed at different ages, and have very different access to primary and specialized care. However, information gathered over the years from individual HGPS case reports, autopsies, and a few clinical trials has helped to define a fairly consistent cardiovascular phenotype. The invaluable HGPS literature prior to the discovery of the genetic cause of the disease (from 1886 to 2003) included patients diagnosed by their characteristic physical appearance, including the so called “old-mannish” look, with a disproportionally big head, baldness, prominent eyes, thin lips, pointed nose, wrinkled and thin skin, and extreme thinness
[7][8]. This allowed the differentiation of HGPS patients from patients with other progeroid-like diseases. Since 2003, HGPS clinical reports include only individuals with confirmed genetic tests, which have identified the classical
LMNA c.1824C>T mutation in most patients (
https://www.progeriaresearch.org/wp-content/uploads/2021/04/FINAL1-PRF-By-the-Numbers_-March-2021.pdf, accessed on March 2021). These recent reports support the previously described HGPS phenotype and have broadened our understanding of HGPS cardiovascular pathology through the use of modern clinical techniques for assessing vasculopathy, such as the measurement of vessel wall echodensity, ankle-brachial index, and pulse wave velocity (PWV). Here, we present a chronological overview of the cardiovascular characterization of HGPS patients that gathers together information from the landmark literature on the clinical definition of HGPS pathology.
2.1. Atherosclerosis and Cardiovascular Calcification
The first thorough anatomical description of a child with remarkable signs of premature aging was reported in 1886 by J. Hutchinson
[9]. A decade later, H. Gilford found that, with no family relationship, Hutchinson’s patient shared a striking physical resemblance to one of his own patients, who looked like an old man, had heart problems, and died at the age of 17
[10]. In the detailed autopsy of this second patient, Gilford noted extensive atheromatous degeneration of the mitral and aortic orifices, obliteration of the lumen of the coronary arteries, soft atheromatous patches on the convex surface of the aortic arch, patches of calcareous material on the concave surface, and extensive atheroma with collections of organized fibrin in the aortic branches
[10].
In 1972, F. L. DeBusk reported four new cases of HGPS and compiled the first thorough list of progeria-like cases from the worldwide literature. In one of his patients, he found enlargement of the right ventricle and a calcified heart valve, and in another he found obstruction of the proximal left coronary artery
[7]. Examination of autopsies revealed that most patients with typical HGPS die of congestive heart failure or acute myocardial infarction due to primary underlying coronary atherosclerosis, and that all patients had varying degrees of generalized atherosclerosis, mainly affecting the large arteries
[7].
In a later review of 12 autopsies, P. B. Baker and colleagues found that cardiac complications in progeria patients were rarely due to cardiomyopathy (interstitial fibrosis in the myocardium without severe coronary artery disease); rather, most cardiac manifestations were due to atherosclerosis-related coronary artery narrowing or occlusion
[11]. Atherosclerosis and calcification of aortic and mitral valves were also common, and all cases featured atherosclerotic plaques in the aorta, ranging from small fatty streaks to complicated calcified lesions. In addition, Baker et al. reported the case of a patient with significant thickening of the intima and media in small cardiac intramural arteries accompanied by diffuse interstitial fibrosis, suggesting small artery disease as a possible cause of cardiac ischemia
[11].
2.2. Vascular Smooth Muscle Cell (VSMC) Loss and Vascular Fibrosis
In 2001, W. E. Stehbens and colleagues reported an unusual histological finding upon reviewing two new cases of HGPS patients who had died of myocardial infarction
[12]. Both patients had severe depletion of VSMCs in the aortic media. The VSMCs were replaced by collagen fibrils, and their depletion correlated with the presence of atherosclerosis and hemodynamic stress around branch sites. The author concluded that the VSMC debris in the fibrosed medial layer indicated muscle degeneration rather than muscle atrophy, and that increased arterial fibrosis may reduce the viscoelastic properties of the wall and contribute to increased blood pressure.
A further analysis in 2006 by R. C. M. Hennekam reported 10 new cases of HGPS and reviewed 132 cases from the literature
[8]. The report covered the range from individual phenotypic features to common symptoms shared by all patients. None of the patients showed signs of a cardiovascular phenotype until around 6 to 8 years of age, when it began to manifest as shortness of breath with exertion and easy fatigability. Other common cardiovascular features included an increase in heart rate and blood pressure with age, heart enlargement and impaired coronary function attributable to an enlarged left ventricle, hypertrophy of myocardial cells and cardiac interstitial fibrosis, angina pectoris appearing up to five years before death, extensive VSMC loss and thickening of the coronary arteries with atheroma plaques (with and without calcification), and thickening of the aortic leaflets, which were often calcified. In addition, half the patients had mitral valve defects, and the aorta had a very variable appearance, from almost normal to severely atherosclerotic. Vascular problems in these patients often extended to the brain, with silent or symptomatic strokes and cerebral infarction frequently reported.
The first prospective clinical characterization of HGPS patients was reported in 2008 by M. A. Merideth et al.
[13]. These authors followed 15 HGPS patients aged between 1 and 17 years over 16 months. Most of the patients had elevated platelet counts, a prolonged prothrombin time, and elevated serum phosphorus. Another frequent feature was the development of age-dependent impaired vascular function, including elevated blood pressure, increased arterial augmentation rate, and reduced ankle-brachial index, together with adventitial thickening. Less frequent abnormalities were thickened aortic valves with regurgitation, left ventricular hypertrophy, pulmonary hypertension, stenotic lesions in the cerebral and carotid arteries, and electrocardiographic alterations that were not associated with ischemia
[13].
In 2010, M. Olive et al. published the first structural and immunohistological comparison of cardiovascular tissues between HGPS and non-HGPS individuals, examining two HGPS patients who died of myocardial infarction and a small non-HGPS cohort composed of 29 individuals with or without CVD with ages ranging from 1 month to 97 years
[14]. Atherosclerosis in HGPS patients and in normally aged individuals shared several features, such as severe stenosis, extensive arterial calcification, and the presence of a range of early- to late-stage atherosclerotic lesions displaying calcification, inflammation, and evidence of erosion or rupture. However, HGPS lesions tended to have smaller atheromatous cores, very large regions of calcification, and thick fibrosis and were categorized predominantly as fibro-calcific lesions and less frequently as fibro-atheromas. Moreover, compared with geriatric vessels, all HGPS vessels (arteries and veins) showed prominent adventitial fibrosis. This study also revealed that progerin is expressed not only in the vessels of HGPS patients, but also in a small subset of cells in the coronary arteries of non-HGPS individuals, with progerin levels increasing with age
[14]. Additional studies have confirmed low progerin expression in several human cell types and tissues
[15][16][17][18][19][20][21][22][23][24][25][26][27][28], but its functional relevance remains to be determined.
2.3. Vascular Stiffening: A Useful Readout for Clinical Trials
Researchers have also investigated whether end-stage cardiovascular events in HGPS are related to progressive impairment of vascular compliance. In a prospective single-center study, M. Gerhard-Herman et al. enrolled 26 HGPS patients and 62 age- and gender-matched healthy children
[29]. All the HGPS patients exhibited vascular stiffness, evidenced by markedly elevated carotid-femoral PWV values comparable to those in adults older than 60 years. This finding was later corroborated in another study
[30]. Other HGPS patient characteristics included higher carotid echobrightness in the intima-media, near the adventitia and deep adventitia, indicating increased arterial wall density and suggesting accumulation of thick collagen fibrils, as well as elevated internal carotid artery mean flow velocity and abnormal ankle-brachial indices, indicating occlusive stenosis and peripheral vascular disease, respectively
[29]. HGPS was therefore classified as a disease of vascular stiffening in the setting of gradual vascular stenosis, similar to CVD in normal aging. Subsequently, PWV and arterial wall echodensity became key cardiovascular readouts of therapeutic efficacy in HGPS clinical trials
[31][32][33]. The first HPGS clinical trial showed improved vascular stiffness upon treatment with the farnesyltransferase inhibitor lonafarnib, from pre-therapy PWV values typical of people aged 60–69 years to end-of-therapy PWV values corresponding to 40–49-year-old individuals
[31]. Moreover, lonafarnib treatment reduced the echobrightness of the carotid intima-media and near/deep adventitia. These findings thus suggested that lonafarnib therapy had the potential to reduce fatal and not-fatal HGPS cardiovascular events and strokes. A second clinical trial showed that triple therapy with lonafarnib plus the statin pravastatin and the bisphosphonate zoledronic acid produced benefits in bone mineral density but did not improve vascular stiffness or structure more than lonafarnib alone
[32]. Instead, the triple therapy resulted in increased development of carotid and femoral arterial plaques and extraskeletal calcifications, thus discouraging recommendation of its use for the clinical treatment of HGPS
[32]. The results from both clinical trials indicated that single lonafarnib therapy can significantly lower mortality rate
[33], and in light of these studies, in November 2020 lonafarnib (marketed as Zokinvy) became the first U.S. Food and Drug Administration-approved drug for HGPS
[34].
2.4. Cardiac Dysfunction
Clinical evaluation of potential treatments for HGPS also requires the definition of appropriate cardiac endpoints. Several electrocardiographic defects have been reported in HGPS patients
[13][30][35], and one defect type frequently found as patients age is cardiac repolarization anomalies
[35][36]. Regarding cardiac anatomy and function, Prakash et al. recently reported that the most frequent echocardiographic abnormality showing increased prevalence with age in HGPS patients is left ventricular (LV) diastolic dysfunction
[30]. The authors speculated that LV dysfunction might be a consequence of myocardial interstitial fibrosis and endocardial thickening, which was observed in two previous case reports
[8][14]. Other cardiac alterations, such as LV hypertrophy and aortic valve calcification and dysfunction, were less common and more frequently seen during the second decade of life
[30]. In line with previous observations in the carotid artery
[29], 70% of HGPS patients had unusually high echobrightness in the aortic root wall, and these patients were more likely to present with diastolic LV dysfunction
[30]. Interestingly, increased echobrightness was not associated with age, suggesting that extracellular matrix (ECM) remodeling may be an early alteration potentially underlying the development of the cardiac phenotype. Another recent study
[37] applied three-dimensional echocardiography (speckled tracking imaging) to analyze seven HGPS patients (five children ≤ 8 years of age and two children in their teens), 21 aged-matched healthy children, and 14 older healthy volunteers (mean age: 65.7 ± 7.5). Despite the fact that most HGPS patients were in their first decade of life, the authors found one case of diastolic dysfunction and another of aortic valve calcification, severe aortic stenosis, and LV hypertrophy, in agreement with some of the findings reported by Prakash et al.
[30].
The data accumulated over several decades indicates that the HGPS cardiovascular phenotype is defined by generalized atherosclerosis with a wide spectrum of early- to late-stage atherosclerotic plaques, prominent VSMC loss, vascular stiffening, calcification and fibrosis, cardiac repolarization abnormalities, LV diastolic dysfunction, and cardiac valve disease, all of which probably contribute to the death of HGPS patients from myocardial infarction, stroke, or heart failure. However, many fundamental questions remain unanswered, and the natural history of CVD leading to these death-causing pathologies is ill defined. Moreover, it remains to be determined whether LV diastolic dysfunction and the altered electrocardiographic activity commonly observed in HGPS patients are early cardiovascular complications or are secondary to other chronic pathologies that precede their onset, such as long-term vascular stiffening and calcification, fibrosis, and ischemia. It is therefore essential to investigate the natural history of HGPS-associated CVD and its underlying mechanisms in order to provide clinicians with the scientific support they need to fight disease progression earlier and more efficiently. The discovery of the genetic mutation causing classical HGPS in 2003
[2][3] triggered the generation of a number of HGPS experimental models that play a pivotal role in the investigation of progerin-induced alterations and the testing of new therapeutic approaches. In the next section we describe the major HGPS-like animal models, with a special emphasis on those that recapitulate aspects of HGPS-associated CVD.
3. An Integrated Model for the Development of HGPS-Associated Cardiovascular Dysfunction
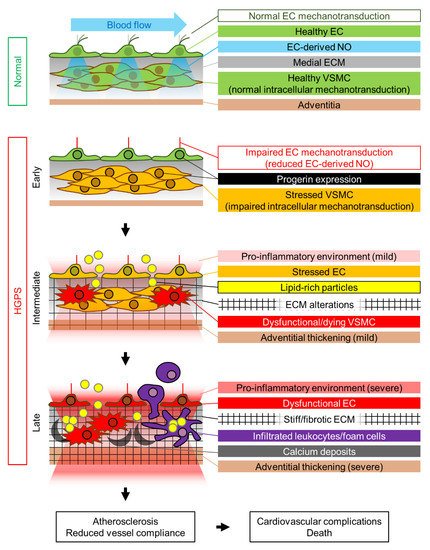
We propose that the HGPS-associated vascular phenotype is due to progerin-induced alteration of multiple mechanotransduction pathways in the vessel wall that affect both VSMCs and ECs (). The pulsatile flow of blood in the arteries subjects the arterial wall to continuous mechanical stress. For reasons that remain elusive, progerin-expressing VSMCs appear to be particularly sensitive to this constant cyclic insult, which induces ER stress, dedifferentiation, calcification (and possibly acquisition of an osteogenic phenotype), DNA damage, and eventually VSMC death, probably due to progerin-mediated altered nucleocytoskeletal connections and defective intracellular force transmission. VSMC damage results in the production of proinflammatory cytokines and ECM remodeling in the arterial adventitial, medial, and probably intimal layers. This proinflammatory environment, together with the alteration of mechanical cues induced by vessel stiffening, likely affects endothelial structure, gene expression, and function. Additionally, progerin-expressing ECs fail to properly mechanotransduce atheroprotective and antifibrotic laminar flow-mediated signals, resulting in decreased eNOS expression. Subsequent reduction in the production of endothelial NO (and probably other angiocrine factors) also contributes to VSMC dedifferentiation and the acquisition of a pro-fibrotic phenotype, thereby establishing a detrimental feedback loop of vascular damage. The reduction in vessel compliance can induce cardiac problems, which may be worsened by the pro-fibrotic environment generated by progerin-expressing cardiac ECs. EC alterations may also further promote atherosclerosis development by increasing endothelial permeability. We propose that the main difference between the mechanisms that regulate atherosclerosis onset in physiological aging and in HGPS is the primary factor inducing endothelial dysfunction. During normal aging, the primary causes of endothelial stress are mainly vessel-extrinsic factors such as dyslipidemia (e.g., hypecholesterolemia, hypertriglyceridemia) and hyperglycemia, which compromise endothelial function and trigger and sustain the vascular damage loop that involves VSMC dysfunction. Conversely, in HGPS patients this cycle is started by VSMC damage, which promotes endothelial dysfunction (). Both scenarios ultimately lead to atherosclerosis development and cardiovascular complications, the main cause of death in both the elderly population and HGPS patients.
Figure 1. Proposed mechanism of HGPS-associated cardiovascular dysfunction. Progerin expression is represented by a black nuclear rim. EC, endothelial cell; ECM, extracellular matrix; NO, nitric oxide; VSMC, vascular smooth muscle cell.
 +1 credit
+1 credit