
When the first cases of HIV infection appeared in the 1980s, AIDS was a deadly disease without any therapeutic alternatives. Currently, there is still no cure for most cases mainly due to the multiple tissues that act as a reservoir for this virus besides the high viral mutagenesis that leads to an antiretroviral drug resistance. Throughout the years, multiple drugs with specific mechanisms of action on distinct targets have been approved.
- HIV
- clinical trials
- novel antiretrovirals
- vaccines
- advanced transdermal nanodelivery systems
1. Introduction
The human immunodeficiency virus (HIV) is still a very prominent disease worldwide. Acquired Immunodeficiency Syndrome (AIDS) can now be considered a chronic infection since patients are living longer due to the several options of antiretroviral therapy [1]. The number of new cases has decreased, but nevertheless, according to the latest UNAIDS global statistics, 38.0 million people are living with HIV in 2019 and 1.7 million became newly infected with HIV in 2019 [1][2].
The initial step of HIV replication cycle involves SU binding to the host protein CD4, present in T helper lymphocytes, macrophages, and dendritic cells. The interaction between the CD4 binding site (CD4 bs) present in SU subunit with CD4 causes conformational changes in the SU glycoprotein that exposes the coreceptor binding site [3][4]. Binding to coreceptor induces further conformational changes in the TM subunit, disclosing the fusion peptide which will connect to the target cell membrane due to its extremely hydrophobic nature, thereby enabling the fusion of both viral envelope and host cell membrane [4]. Fusion of viral envelope could occur directly with plasma membrane or alternatively with endosome membrane after endocytosis. Following fusion, the viral capsid is then released in the cytosol forming the reverse transcriptase/pre-integration complex (RTC/PIC) that includes the two copies of gRNA, the viral proteins CA, NC, IN, RT, and Vpr as well as cellular proteins, namely cyclophilin A [5]. Inside this structure, RT converts the single-stranded gRNA into a double stranded DNA (dsDNA). The RTC/PIC is transported through the cytoplasm into the host cell nucleus [5][6] via the nuclear pore, where RTC/PIC interacts with proteins of the nuclear pore complex, namely, Nucleoporin 358 and Nucleoporin153 [5][7]. The structural integrity of RTC/PIC seems to be maintained until it enters the nucleus, minutes before the dsDNA integrates into host cell chromosome [5][6]. The integration process is mediated by the integrase protein (IN) starting by removing nucleotides from the 3′ ends of the proviral DNA, and then, proceeding to catalyze a nucleophilic attack to the phosphodiester bonds of the DNA chains, thus forming a covalent bond between viral and host DNA. This is an essential step in viral replication allowing the establishment of latently infected cells [8][9][10]. Some cell transcription factors enhancers bind to LTR and the regulatory proteins such as Tat, Rev, and Nef are produced. HIV-1 transcription from the LTR promoter is activated by the Tat protein through interaction with the nascent trans -acting-responsive RNA hairpin structure [9][10]. The Env glycoproteins, after translation, processing, and cleavage by cellular furin protease migrate to the plasma membrane. Meanwhile the MA domain targets the Gag-Pol polypeptide to budding sites at the host cell membrane where it interacts with cytoplasmic tail of TM subunit of Env. This event allows virion assembly with gRNA and all the other structural and accessory viral proteins and budding of immature viral particles [11]. During or soon after budding, the viral protease (PR) cleaves the Gag-Pol polypeptide which will allow the release of all structural proteins, such as MA, CA, and NC, as well as their reorganization into mature virions [9][10].
The period between the infection of the first host cell and the detection of the virus in the blood is called the eclipse phase and usually lasts from 7 to 21 days. After the infection of the first cell, the virus continues to replicate in the mucosa, submucosa, and adjacent lymphatic tissue. The replication concentrates in the gut-associated lymphatic tissue (GALT) quite early [12][13]. Then, it follows an exponential rise of the viral loading in which the CD4+ cell counting rapidly decreases. This phase is characterized by flu-like and non-specific clinical signs that usually last between 7 to 10 days. After a few weeks, the immune system can generate a response [9][14]. The cellular immune response starts with the activation of CD8+ cytotoxic lymphocytes. Their T-cell receptor (TCR) will bind to viral proteins, which are in turn connected to the antigen-presenting molecule (MHC I) to eliminate the infected cells [9]. Generally, after 3 to 5 weeks, a humoral response starts to produce specific neutralizing antibodies that will destroy the virions via phagocytosis. The convergence of both types of immune responses leads to a decrease in viremia and a new rise in CD4+ cell count. The period, in which there is an infection but without antibodies, is called the “serological window period” [14]. Even though this is an asymptomatic phase, and the viral loading is somewhat controlled, there is still a loss of immune cells since the virus continues to replicate in the lymphatic tissue (its reservoir), destroying its structure. Then, the viral loading becomes higher as the CD4+ cell count diminishes, leading to the beginning of the AIDS stage. In this stage, the patients are more susceptible to opportunistic infections [14].
Several aspects make it difficult to eradicate the virus once a patient is infected. One of them is related to the absence of proofreading activity in the viral RT, causing a great number of mutations and genetic diversity in the HIV genome. The other one is concerned with the ability of the virus to infect resting memory or naïve cells, leading to a latent viral state [9][14]. The problem with viral latency is that it can occur even after patients have undergone antiretroviral therapy reducing viremia to an undetectable level [9].
2. Novel Therapeutic Strategies
2.1. New Transdermal Drug Delivery Systems
2.1.1. Transdermal Delivery System for Tenofovir Alafenamide
2.1.2. Transdermal Delivery of Enfuvirtide (T20) via Ultrasounds
2.2. Nanosystems for Drug Delivery
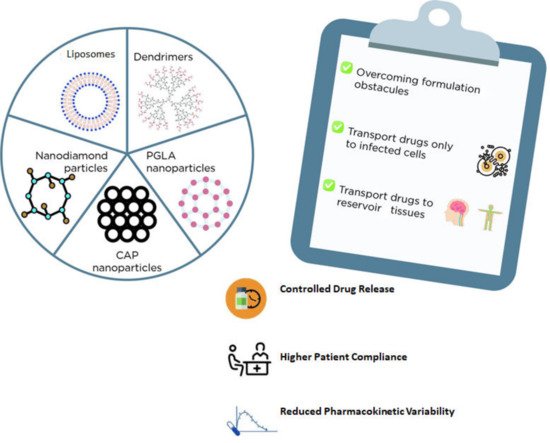
| Nanocarriers + ARV | Main Outcomes | References |
|---|---|---|
| Liposomes + Stavudine | Liposomes were revealed to be a promising alternative for stavudine delivery as these carriers can be easily absorbed by macrophages. | [24] |
| Dendrimer + Zidovudine | The formulation reduced the AZT hemolytic effect and prolonged the drug release, decreasing the occurrence of side effects. | [26] |
| Carbosilane Dendrimers+Zidovudine Carbosilane Dendrimers + Efavirenz Carbosilane Dendrimers + Tenofovir |
An enlarged antiviral activity of all three drugs was observed when formulated with dendrimers. | [27] |
| Nanodiamond Particles + Efavirenz | A suitable and slower release profile through a blood–brain barrier model was obtained impairing viral replication for a longer period. | [30] |
| PGLA nanoparticles + Efavirenz PGLA nanoparticles + Saquinavir |
An enlarged antiviral activity of all three drugs was obtained with PGLA nanoparticles. | [31] |
| PGLA nanoparticles + Efavirenz + Raltegravir [thermosensitive gel] | A lower EC90 and a constant release of these loaded drugs were obtained being a promising option for pre-exposure HIV prophylaxis. | [32] |
| CAP nanoparticles + Efavirenz (thermosensitive gel) |
High encapsulation efficacy and lower cytotoxicity in HeLa cells were observed besides enhanced prophylactic activity in TMZ-bl cells treated with EFV-CAP nanoparticles. | [34] |
| CAP nanoparticles + Dolutegravir (thermosensitive gel) |
pH (4.2 and 7.4) influenced both the drug release and the cytotoxicity of this formulation. | [33] |
References
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271.
- UNAIDS. Global HIV & AIDS Statistics—2019 Fact Sheet. UNAIDS. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 10 November 2019).
- German Advisory Committee Blood (Arbeitskreis Blut), Subgroup ‘Assessment of Pathogens Transmissible by Blood’ GACB (Arbeitskreis, Blood’ S ‘Assessment of PT by. Human immunodeficiency virus (HIV). Transfus. Med. Hemother. 2016, 43, 203–222.
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866.
- Schaller, T.; Ocwieja, K.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hué, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 Capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7, e1002439.
- Li, C.; Burdick, R.C.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc. Natl. Acad. Sci. USA 2021, 118, e2019467118.
- Toccafondi, E.; Lener, D.; Negroni, M. HIV-1 capsid core: A bullet to the heart of the target cell. Front. Microbiol. 2021, 12, 755.
- Sierra, S.; Kupfer, B.; Kaiser, R. Basics of the virology of HIV-1 and its replication. J. Clin. Virol. 2005, 34, 233–244.
- Krogstad, P. Molecular biology of the human immunodeficiency virus: Current and future targets for intervention. Semin. Pediatr. Infect. Dis. 2003, 14, 258–268.
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Genet. 2012, 10, 279–290.
- Meng, B.; Lever, A.M. Wrapping up the bad news—HIV assembly and release. Retrovirology 2013, 10, 5.
- Shaw, G.M.; Hunter, E. HIV transmission. Cold Spring Harb. Perspect. Med. 2012, 2, a006965.
- Cohen, M.S.; Shaw, G.M.; McMichael, A.J.; Haynes, B.F. Acute HIV-1 infection. N. Engl. J. Med. 2011, 364, 1943–1954.
- Fanales-Belasio, E.; Raimundo, M.; Suligoi, B.; Buttò, S. HIV virology and pathogenetic mechanisms of infection: A brief overview. Ann. Ist. Super Sanita. 2010, 46, 5–14.
- Singh, N.; Singh, R.; Verma, V. An Introduction to the Transdermal Delivery of Antiretrovirals. Adv. Biol. BioMed. 2014, 1, 1–32, Article ID: BIO14 06.
- Ham, A.S.; Buckheit, R.W. Current and emerging formulation strategies for the effective transdermal delivery of HIV inhibitors. Ther. Deliv. 2015, 6, 217–229.
- Puri, A.; Bhattaccharjee, S.A.; Zhang, W.; Clark, M.; Singh, O.N.; Doncel, G.F.; Banga, A.K. Development of a transdermal delivery system for tenofovir alafenamide, a prodrug of tenofovir with potent antiviral activity against HIV and HBV. Pharmaceutics 2019, 11, 173.
- Vedha Hari, B.N.; Devendharan, K.; Narayanan, N. Approaches of novel drug delivery systems for Anti-HIV agents. Int. J. Drug. Dev. Res. 2013, 5, 16–24.
- Snook, K.A.; Van Ess, R.; Werner, J.R.; Clement, R.S.; Ocon-Grove, O.M.; Dodds, J.W.; Ryan, K.J.; Acosta, E.P.; Zurlo, J.J.; Mulvihill, M.L. Transdermal delivery of enfuvirtide in a porcine model using a low-frequency, low-power ultrasound transducer patch. Ultrasound Med. Biol. 2019, 45, 513–525.
- Tachibana, K.; Tachibana, S. The use of ultrasound for drug delivery. Echocardiography 2001, 18, 323–328.
- Luis, J.; Park, E.J.; Meyer, R.J.; Smith, N.B. Rectangular cymbal arrays for improved ultrasonic transdermal insulin delivery. J. Acoust. Soc. Am. 2007, 122, 2022.
- Marwah, H.; Garg, T.; Goyal, A.K.; Rath, G. Permeation enhancer strategies in transdermal drug delivery. Drug Deliv. 2014, 23, 564–578.
- Grande, F.; Ioele, G.; Occhiuzzi, M.A.; De Luca, M.; Mazzotta, E.; Ragno, G.; Garofalo, A.; Muzzalupo, R. Reverse transcriptase inhibitors nanosystems de-signed for drug stability and controlled delivery. Pharmaceutics 2019, 11, 1–26.
- Nayak, D.; Boxi, A.; Ashe, S.; Thathapudi, N.C.; Nayak, B. Stavudine loaded gelatin liposomes for HIV therapy: Preparation, characterization and in vitro cytotoxic evaluation. Mater. Sci. Eng. C 2017, 73, 406–416.
- Mhlwatika, Z.; Aderibigbe, B.A. Application of dendrimers for the treatment of infectious diseases. Molecules 2018, 23, 2205.
- Kumar, S. In-vitro and in-vivo evaluation of poly (propyl ether imine) (petim) dendrimer for sustained delivery of zidov-udine. J. Antivir. Antiretrovir. 2013, S10, 1–7.
- Vacas-Córdoba, E.; Galán, M.; de la Mata, F.J.; Gómez, R.; Pion, M.; Muñoz-Fernández, M.Á. Enhanced activity of carbosilane den-drimers against HIV when combined with reverse transcriptase inhibitor drugs: Searching for more potent microbicides. Int. J. Nanomed. 2014, 9, 3591–3600.
- Sarmento, B.; Gomes, M.J.; das Neves, J. Nanoparticle-based drug delivery to improve the efficacy of antiretroviral therapy in the central nervous system. Int. J. Nanomed. 2014, 9, 1757–1769.
- Curley, P.; Liptrott, N.J.; Owen, A. Advances in nanomedicine drug delivery applications for HIV therapy. Futur. Sci. OA 2018, 4, FSO230.
- Roy, U.; Drozd, V.; Durygin, A.; Rodriguez, J.; Barber, P.; Atluri, V.; Liu, X.; Voss, T.G.; Saxena, S.; Nair, M. Characterization of nanodiamond-based anti-HIV drug delivery to the brain. Sci. Rep. 2018, 8, 1–12.
- Chaowanachan, T.; Krogstad, E.; Ball, C.; Woodrow, K.A. Drug synergy of tenofovir and nanoparticle-based antiretrovirals for HIV prophylaxis. PLoS ONE 2013, 8, e61416.
- Date, A.; Shibata, A.; Goede, M.; Sanford, B.; La Bruzzo, K.; Belshan, M.; Destache, C.J. Development and evaluation of a thermosensitive vaginal gel containing raltegravir+efavirenz loaded nanoparticles for HIV prophylaxis. Antivir. Res. 2012, 96, 430–436.
- Mandal, S.; Khandalavala, K.; Pham, R.; Bruck, P.; Varghese, M.; Kochvar, A.; Monaco, A.; Prathipati, P.K.; Destache, C.; Shibata, A. Cellulose acetate phthalate and antiretroviral nanoparticle fabrications for HIV pre-exposure prophylaxis. Polymers 2017, 9, 423.
- Date, A.A.; Shibata, A.; Mcmullen, E.; La Bruzzo, K.; Bruck, P.; Belshan, M.; Zhou, Y.; Destache, C.J. Thermosensitive gel containing cellulose acetate phthalate-efavirenz combination nanoparticles for prevention of HIV-1 infection. J. Biomed. Nanotechnol. 2015, 11, 416–427.
- Pons-Faudoa, F.P.; Sizovs, A.; Shelton, K.A.; Momin, Z.; Niles, J.A.; Bushman, L.R.; Xu, J.; Chua, C.Y.X.; Nichols, J.E.; Demaria, S.; et al. Preventive efficacy of a tenofovir alafenamide fumarate nanofluidic implant in SHIV-challenged nonhuman primates. Adv Ther (Weinh). 2021, 4, 2000163.