Your browser does not fully support modern features. Please upgrade for a smoother experience.
TP53 Pathway in Embryonic/Somatic Cells: History

Subjects:
Pathology
The P53 pathway is the most important cellular pathway to maintain genomic and cellular integrity, both in embryonic and non-embryonic cells. Stress signals induce its activation, initiating autophagy or cell cycle arrest to enable DNA repair.
- P53 pathway
- cancers
- mutations
- embryonic and somatic cells
1. Embryonic Stem Cells and Germ Cell Tumors Versus Somatic Cells
The development of multicellular, sexually reproductive organisms starts with the fusion of a spermatozoa and an oocyte that created a diploid zygote (depicted in Figure 1). Then, the zygote divides, forming a cluster of undifferentiated cells (i.e., blastomeres) known as the morula. The first step of lineage differentiation occurs during the subsequent blastocyst stage in which the embryoblast (i.e., the inner cell mass) and the trophectoderm are formed [1,2]. The trophectoderm develops into all extra-embryonic structures, whereas the embryoblast, which consists of pluripotent embryonic stem (ES) cells, develops into all embryonic structures [2]. ES cells are transiently present, can self-renew and give rise to all three embryonic germ layers during gastrulation (i.e., the ecto-, meso-, and endoderm), which ultimately will give rise to all cell lineages of the adult organism [2]. Additionally, ES cells give rise to the primordial germ cells (PGC) in the embryonic yolk sac, which subsequently migrate toward developing gonads to give rise to the germ line [2,3] (see Figure 1).
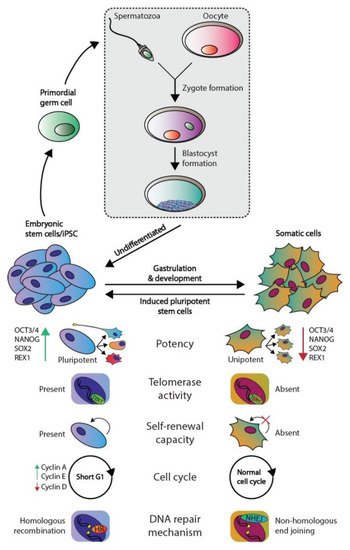
Figure 1. Phenotypic differences between embryonic and somatic cells. Fertilization of an oocyte by a spermatozoon forms a zygote, which subsequently develops the blastocyst. The blastocyst contains embryonic stem (ES) cells that eventually give rise to all somatic cell types. ES and somatic cells can be distinguished according to multiple aspects: their potency, telomerase activity, self-renewal capacity, cell cycle regulation, and preferred DNA repair mechanism. Both germ cells and germ cells tumors are derived from ES derived PGCs and therefore harbor the same embryonal characteristics.
The pluripotent nature of ES cells is characterized by the expression of pluripotency markers including but not limited to OCT4, NANOG, SOX2, and REX1 (Figure 1) [4]. In contrast, somatic cells are differentiated and thus lineage-restricted and unipotent [5]. Therefore, these cells seldomly give rise to other progeny than the identity of the cells themselves [5]. In addition, due to their capacity to self-renew, ES cells can be cultured indefinitely in vitro while retaining their embryonic state as well as a stable genome. This immortality is facilitated by active telomerase, which is an enzyme that extends telomeric repeats that are otherwise lost due to the end-replication problem after each cell cycle (approximately 50–150 base pairs are lost per cycle) [6,7] (Figure 1). Conversely, somatic cells have a restricted lifespan due to the lack of active telomerase and induce cellular senescence when a critical telomere length is reached [6,7]. Furthermore, ES cells also regulate the cell cycle differently as these lack Cyclin D expression yet continuously express Cyclin A and E, which leads to a significantly shortened G1-phase when compared to somatic cells [8]. This characteristic also facilitates the tendency for ES cells to initiate apoptosis rather than cell cycle arrest and DNA repair when DNA damage occurs, the latter of which occurs predominantly in the G1 phase after cell cycle arrest [9,10]. The preference for apoptosis in these early embryonic cells is considered a failsafe mechanism to preserve the genetic integrity of their multitudinous progeny. In rare occasions, ES cells can initiate DNA repair; however, these cells then opt for error-free homologous recombination (HR) to repair double-strand breaks, whereas somatic cells predominantly employ error-prone non-homologous end joining (NHEJ) [9]. Finally, a zygote is known to lose its inherited DNA methylation pattern immediately after fertilization and subsequent changes in DNA methylation occur during early embryonic development as various cell lineages arise. Thus, DNA methylation patterns between ES and somatic cells vary significantly [11,12].
Of note, a discovery made over a decade ago demonstrated that differentiated somatic human cells (e.g., adult human fibroblasts) could be reprogrammed to a pluripotent state reminiscent of ES cells (i.e., nuclear reprogramming) [13]. These cells are known as induced pluripotent stem cells (iPSCs) and, in theory, they can also be maintained in a pluripotent state indefinitely. iPSCs are originally generated in vitro by overexpressing a cocktail of four essential genes (OCT3/4, c-MYC, SOX2, and KLF4) in the differentiated cells [14].
Moreover, many of the characteristics of ES cells are strongly conserved in germ cell tumors (GCTs), which represent a heterogeneous cluster of solid tumors that originate from both ES cells and PGCs [3]. Of note, the parallels observed between ES cells and GCTs do not apply to the teratomas, which is a GCT subtype that consists of fully differentiated cells and harbor significant differences in their (epi)genetic makeup and clinical response (i.e., inherent chemotherapeutic resistance) compared to other GCT subtypes [3,15,16,17]. An example of the similarities observed between ES cells and GCTs is that the latter also express multiple pluripotency markers including OCT3/4 and NANOG and have active telomerase activity, again with the exception of teratoma [3]. Moreover, it is widely accepted that in response to DNA damage, GCTs also favor apoptosis, which is thought to underlie their unique sensitivity to platinum-based chemotherapeutics, including cisplatin (a key component of GCT treatment) [9,10,18]. This is supported by additional evidence demonstrating that GCTs have low expression of genes involved in cell cycle arrest and a deregulated G1-S phase checkpoint, thus potentially also preventing the activation of DNA repair pathways [18,19,20,21,22,23]. Moreover, the apoptotic response of ES cells and GCTs in response to DNA damage has been attributed to the well-known P53 pathway; however, at least in GCTs, it remains a topic of debate [18,21,24,25,26,27,28,29,30,31,32,33,34,35,36,37]. The P53 pathway has been dubbed the guardian of the genome due to its integral role in maintaining genomic integrity among all cell types. This pathway acts in response to cellular stress and lies at the apex of a plethora of downstream signaling pathways including cell cycle arrest, DNA repair, and apoptosis. The deregulation of this pathway has been observed in cancer and is strongly associated to many aspects of tumorigenesis. For instance, GCTs most often express high levels of wild-type (WT) TP53, and it has been suggested that the P53 pathway is (partially) responsible for their characteristic to enter apoptosis in response to platinum-based chemotherapeutics (e.g., cisplatin) which is reminiscent of ES cells in response to DNA damage [10,18,21,23,25,26,27,28,29,33,34,35,36,37]. In addition, it has been suggested that the P53 pathway is deregulated through multiple mechanisms in GCTs that have acquired resistance to chemotherapy, including cisplatin [15,18,38,39,40,41,42].
2. Protecting Genomic Integrity
Genomic changes during development serve as the driving force of evolution; however, these may also negatively impact multiple cell lineages with possible detrimental effects on both the individual and their offspring. Therefore, protection of the genomic integrity is paramount for a species to survive and commences during early embryonic development [23]. For example, the oocyte expresses and deposits multiple mRNAs that protect the zygote against DNA damage during the first cellular divisions [43]. As mentioned before, there have been several indications of a robust protective mechanism in ES cells including the removal of mutated ES cells from the stem cell pool through apoptosis, which is a characteristic that is seemingly conserved in GCTs [7,12,44]. Alternatively, ES cells may also be instructed to differentiate through transcriptional inhibition of the pluripotency factor NANOG, which also effectively removes ES cells from the stem cell pool [45]. These mechanisms in combination with lacking a G1 checkpoint are thought to result in a 100-fold lower frequency of accumulating mutations in at least murine ES cells compared to mouse embryonic fibroblasts, which resemble the somatic cells [9,23]. Conversely to ES cells and GCTs, somatic cells utilize different pathways (DNA repair rather than apoptosis) to protect their genome that enable these to survive while risking an increased mutational load [9,23].
3. P53 Pathway
As mentioned before, the P53 pathway is essential in maintaining genomic integrity. Central to this pathway is the P53 protein that is regarded as a tumor suppressor and originates from the TP53 gene. Several key discoveries regarding P53 are depicted in Figure 2, as well as its relevance in GCTs [22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,46,47,48,49,50,51,52,53,54,55,56,57,58,59]. In response to cellular stress, the P53 protein is activated and accumulates in the cell after which it mainly functions as a transcription factor that transactivates a plethora of downstream targets [49]. These target genes are key players in one of many downstream pathways including cell cycle arrest, DNA repair, apoptosis, senescence, and autophagy [51,60,61]. The P53 protein counts 393 amino acids (AA) and consists of multiple domains, starting with a transactivation domain (TAD) at the amino-terminus (N-terminus), a directly neighboring proline-rich domain (PRD), a large DNA-binding domain (DBD), a tetramerization domain (TD), and a carboxy-terminus (C-terminus) regulatory domain (REG) [62,63] (depicted in Figure 3). Once activated and accumulated in the cell, the P53 protein functions and binds DNA as a tetramer, which is facilitated by the TD [64]. When tetramerized, the DBD contains three loops, L1, L2, and L3 respectively [65]. Both L2 and L3 are bound by a zinc ion, effectively linking both loops and enabling L3 to bind to the minor groove of the DNA, while a helix in the DBD is complementary to the major groove [65,66].
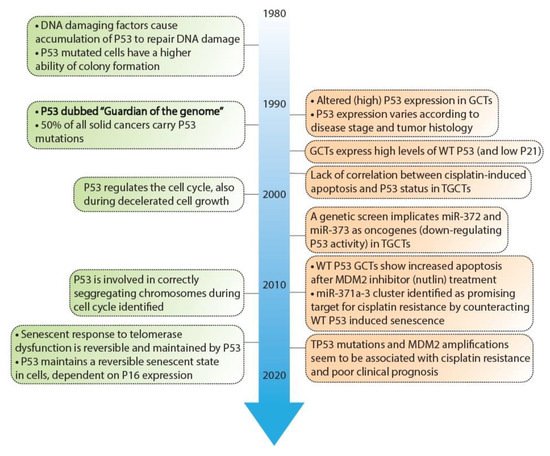
Figure 2. Timeline depicting key discoveries in P53 signaling and germ cell tumors. A timeline of the discoveries regarding P53 as a guardian of the genome (green) and studies regarding TP53 in GCT development and resistance (orange). References key discoveries P53 (green): 49–58, references key discoveries TP53 in GCT development (orange): 25–41, 59–62.

Figure 3. Schematic overview of the P53 protein. Indicated are (from N- to T-terminus) TAD: Transactivation domain, PRD: Proline-rich domain, DBD: DNA-binding domain, TD: Tetramerization domain and REG: Regulatory domain. Numbers indicate the amino acids included in each domain.
There are many different forms of cellular stress such as DNA damage that activate upstream regulators of p53, leading to the activation of the p53 pathway. For example, Ataxia-telangiectasia mutated (ATM) kinase serves as an important DNA damage sensor, as it recognizes and binds to double-strand DNA breaks and subsequently activates the P53 pathway, ultimately resulting in the activation of NHEJ or HR DNA repair mechanisms [55,67]. ATM phosphorylates histone H2AX, which is a variant of histone H2A that recruits molecules responsible for H3K9 methylation [55]. In turn, methylated H3K9 leads to the acetylation and activation of ATM [55]. Among other substrates, ATM phosphorylates and activates Checkpoint Kinase 1 and 2 (CHK1 and CHK2, respectively) [55]. In turn, CHK1 and 2 function as transcriptional activators by phosphorylating and activating the P53 protein [68]. The subsequent section will further outline several downstream targets and their related pathways including cell cycle arrest, senescence, apoptosis, and autophagy (also summarized in Figure 4).
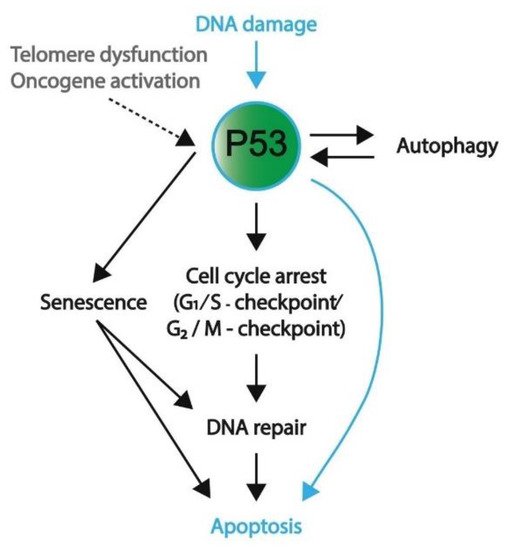
Figure 4. A schematic overview representing the favored mechanism of P53 activation in embryonic cells and somatic cells. DNA damage, both in embryonic (blue) and somatic (black) cells, will induce P53 activation. To protect genomic integrity, embryonic cells favor apoptosis over DNA repair. Conversely, somatic cells preferentially engage in cell cycle arrest and DNA repair; however, if DNA damage persists, these cells will enter apoptosis or become senescent. Furthermore, telomere dysfunction and oncogene activation are other means to activate P53 signaling (mostly in somatic cells). In addition to apoptosis, P53 activation and its downstream pathways can result in autophagy, senescence, and cell cycle arrest.
3.1. Induction of Cell Cycle Arrest
As indicated, DNA damage leads to the accumulation of P53, which induces cell cycle arrest, and subsequent DNA repair cell cycle arrest can occur at both the G1/S and G2/M checkpoints and is mostly effectuated by P21 (CDKN1A), which is a target of P53 [69]. Activated P53 binds to the promotor of P21 and initiates its transcription [70,71]. P21 inhibits the function of cyclin-dependent kinases (CDKs) CDK2 and CDK4/6 present at the G1/S transition, which results in the hypomethylation of pRB-related proteins p107 and p130 [69,72]. The transcription of cell cycle-promoting genes will subsequently be repressed, inducing a temporary block of cell cycle arrest that enables the cell to initiate DNA repair mechanisms.
3.2. Senescence or Apoptosis as a Final Measure to Protect Genomic Integrity
Chronic cellular stress signals such as telomere dysfunction, persisting DNA damage, and oncogene activation result in prolonged P53 activation and subsequent P21 expression, which in turn may induce cellular senescence or apoptosis (the latter of which is the main outcome in ES cells and GCTs) [72]. In contrast to cell cycle arrest, senescence is a stable state of arrest, and both processes are initiated by P53, again illustrating the tumor-suppressive functions of this protein [55,72]. Conversely, a senescent cell is able to re-enter the cell cycle after loss of P53 and has been shown to contribute to tumorigenesis, as it results in fast cell proliferation [27,28,73]. Prolonged P21 expression eventually induces the expression of P16INK16A (hereafter referred to as P16), which similarly functions as a CDK inhibitor for CDK4/6, thereby indirectly inhibiting pRb-family members [72]. P16 expression is known to be regulated by epigenetic factors including DNMT3, which facilitates de novo methylation of the P16 promotor, inhibiting its expression, which is maintained by DNMT1 [55]. Demethylation of the promotor consequently induces P16 expression [55]. In addition to P16, senescence is also regulated by P53-independent pathways including NF-κb, which is responsible for the expression of proinflammatory cytokines that are secreted during senescence/this cellular state [55,72]. Notably, senescent cells are non-responsive to apoptotic signals, which is demonstrated by the continued activation of cAMP response element-binding (CREB), which functions as a transcription factor for the anti-apoptotic BCL-2 protein [55]. Alternatively, persisting cellular stress signals may induce apoptotic pathways, and further information of these pathways is described in Box 1.
3.3. Autophagy
Lastly, P53 can induce autophagy, which underlines its additional role in cellular metabolism. Autophagy enables cells to maintain homeostasis by the removal and recycling of (faulty) organelles [74]. Recycling results in an increase of anabolic intermediates that can subsequently be used in multiple pathways [74,75]. There are many connections between P53 and autophagy, as illustrated in Figure 5, as autophagy is able to inactivate P53 through different mechanisms such as proteosomal degradation [76]. In addition, the autophagy protein ATG7 has been reported to bind P53 and activate the transcription of P21, resulting in cell cycle arrest [74]. Conversely, P53 promotes the transcription of genes involved in autophagy, which suggests a negative feedback loop between the autophagy proteins and P53 [74]. Additionally, a more indirect crosstalk between p53 and autophagy via the mTOR pathway is known to occur in multiple tissue types including skeletal muscle, heart, white fat, liver, and kidney tissue [33]. Here, P53 promotes the transcription of negative regulators of the IGF–AKT–mTOR pathway, resulting in higher levels of BECLIN-1, which is part of the phosphatidyl inositol-3 kinase (PI3K) complex and plays an important role in the formation of autophagosomes [77,78]. Notably, a loss of BECLIN-1 results in an increased risk of tumorigenesis, which underlines its importance in autophagy and maintaining genomic integrity [78].
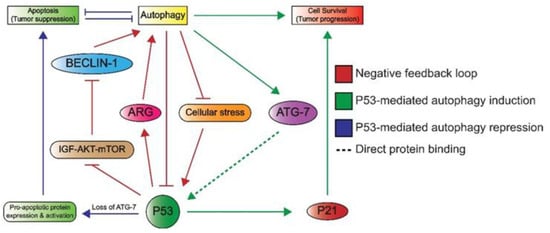
Figure 5. An overview of the crosstalk between P53 and autophagy. Autophagy can repress P53 through the inhibition of upstream activating proteins as well as P53 itself. This forms a negative feedback loop (red) as P53 induces autophagy through the inhibition of the IGF–AKT–mTOR pathway and subsequent increase in BECLIN-1 levels, which plays a role in autophagosome formation. P53 also induces the expression of autophagy-related genes (ARG). Autophagy induces cell survival, which is effectuated by the binding of ATG7 to P53, ultimately leading to P53-dependent P21 expression and subsequent cell cycle arrest (green). The loss of ATG-7 results in apoptosis (blue), as P53 induces the expression and activation of pro-apoptotic proteins. Apoptosis and autophagy have mutually inhibitory effects on one another.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22105377