Your browser does not fully support modern features. Please upgrade for a smoother experience.
Subjects:
Cell Biology
The impact of cell fusion on biology and genetics has been of a subject of interest for many years (see [1,2] for review). Cell fusion occurs in normal placenta, skeletal muscle, and bone [3,4,5]. Cell fusion also occurs in response to injury in liver, heart, and interstitial tissues; in various viral and bacterial infections; and, quite often, in cancer. Of the various conditions in which cell fusion is observed, none is more controversial and potentially more important than malignancy.
Numerous observations in experimental animals and some human subjects suggest that fusion of one normal cell with another can induce malignant transformation, and fusion of a cancer cell with other cells can spark the progression of an existing malignancy to a more malignant phenotype (Figure 1A). However, there also exist observations in various and disparate fields of inquiry potentially connecting cell fusion with natural defenses against malignancy (Figure 1B). How cell fusion could pose a barrier to malignancy and progression of malignancy has not been systematically explored. We do so here.
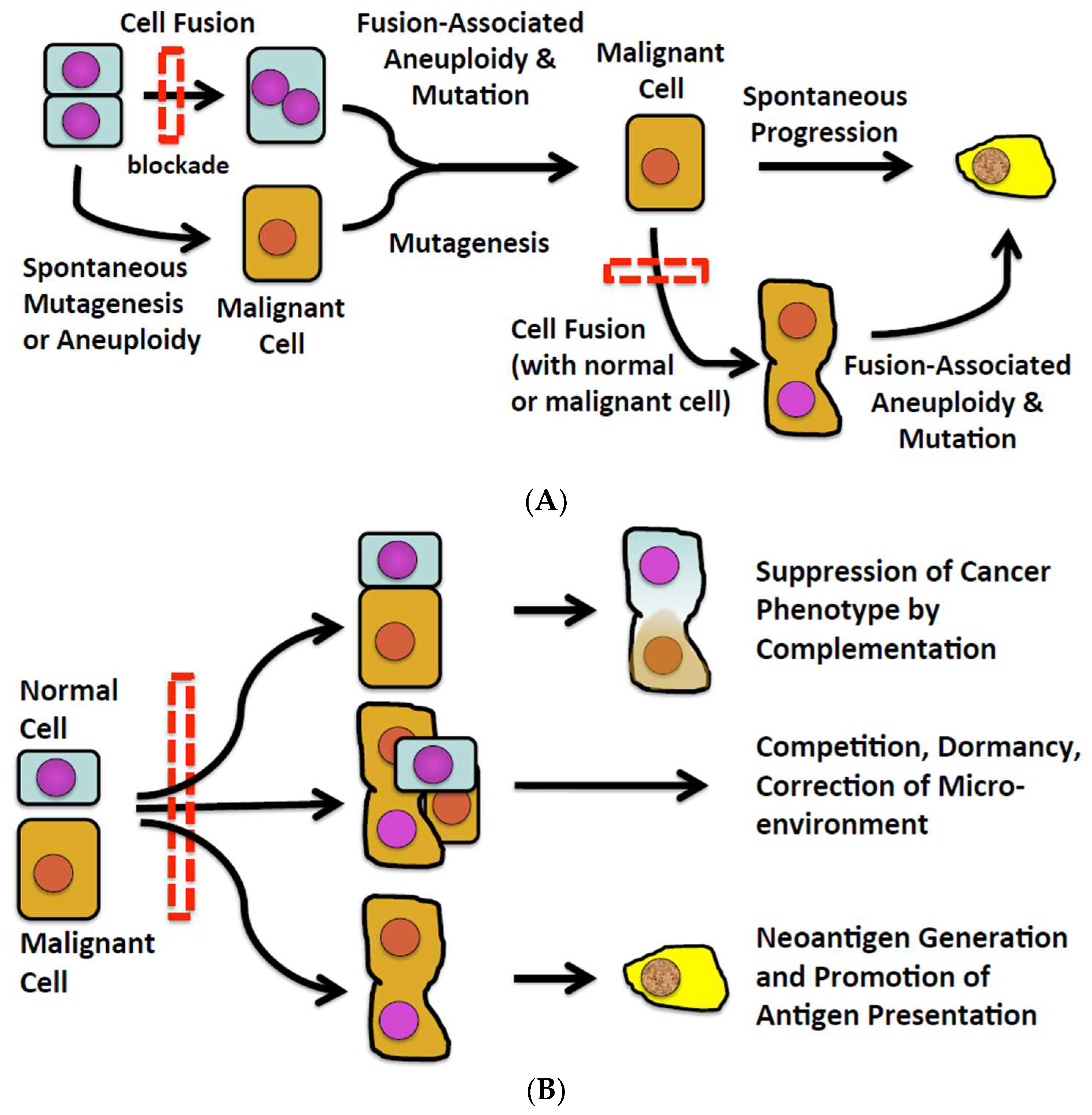
Figure 1. (A) The conventional view of cell fusion in malignancy. (B) A new perspective of cell fusion as a defense against malignancy.
Cell fusion has been detected in some human and animal malignancies (for review, see [2,6,7,8,9,10,11,12,13]), however the process of fusion is rarely, if ever, observed and therefore the prior fusion of a cell must be inferred, usually by analysis of karyotype or complementation in chimeric individuals. Fusion of cancer cells with normal cells in experimental systems can diversify the genome and phenotypes of malignant cells and promote progression [7,10,14,15,16,17,18,19,20]. How often cell fusion does so in naturally arising human malignancies is unknown because few, if any, markers besides the presence of multiple nuclei reliably establish a given cell to be a hybrid. Perhaps the most compelling evidence of cell fusion in cancers is found in hematopoietic chimeras. As one recent example, LaBerge et al. [21] reported that malignant mononuclear cells isolated from a melanoma and metastases of the melanoma in a bone marrow transplant recipient contained DNA from the bone marrow transplant donor and the recipient. Although the process leading to cell fusion was not witnessed, the presence of substantial amounts of donor and recipient DNA in mononuclear hybrid cells most likely reflected spontaneous fusion of donor and recipient cells.
Thus, important questions concerning cell fusion in cancer have long persisted. These questions include: (i) how often cell fusion actually occurs at the inception or after development of cancer; (ii) whether cell fusion causes or results from malignant transformation; (iii) whether cell fusion promotes progression of malignancy or progression promotes cell fusion; (iv) whether cell fusion promotes resistance to malignant transformation or progression of malignancy; and (v) if cell fusion does protect against malignancy and/or progression, whether surviving hybrid tumor cells reflect the failure of protective mechanisms. These questions have been of theoretical interest for many years [1,14,22,23]. Inextricable from these questions are controversies about the mechanisms underlying genetic and chromosomal changes in cancer, especially whether malignancies arise by stepwise accumulation of point mutations or chromosomal damage, aneuploidy, and/or chromosomal instability [24].
These questions occurred to us as we considered the implications of our own work and work conducted by others over time. We found that deliberate fusion of normal epithelial cells can induce malignant transformation and progression of malignancy that ensues [24]. Because fused cells were cloned, it was possible to determine the frequency of transformation and progression in relation to the fusion event. On the other hand, when observed as a spontaneous event in a heterologous system, cell fusion occurred frequently but never eventuated in evident malignancy [25]. There are good reasons to propose that malignant transformation might have occurred in the system in which we observed spontaneous cell fusion (i.e., human hematopoietic stem cells engrafted in fetal swine). However, it is also possible that spontaneous fusion of cells induces protections as well as genetic changes and that the latter dominate in most instances. As we consider the existing set of clinical and experimental observations, it is evident that because fusion events cannot be witnessed, further investigation of human cancers and basic cell biology will offer only provisional answers to the questions posed above. However, the development and testing of agents that specifically and effectively block cell fusion could dramatically advance understanding and perhaps answer all of the questions of long standing about cell fusion in cancer. Therefore, as we discuss observations suggesting cell fusion could promote resistance to malignancy, we shall also consider how specific blockade of cell fusion would modify such resistance.
Therapeutic agents that specifically and effectively inhibit cell fusion do not as yet exist but are likely to emerge. Investigations connecting viral or cellular fusogens with cancers [4,26,27] make fusogens and related molecules attractive targets for therapeutics [28]. Reports connecting cell fusion with the inception and progression of malignancy, such as a report by Duelli et al. [29] using bulk populations of cells and our report on clonal populations [24], might be taken to suggest that inhibition of cell fusion would impact favorably. Consistent with that concept, blockade of IL-4 receptors, the ligation of which induces fusion of myoblasts, suppresses the initiation and progression of alveolar rhabdomyosarcoma [30], and digestion of syntaxin 1 with botulinum toxin C1 decreases progression of human glioblastoma cells in immunodeficient mice, as only two examples. While consistent with the importance of cell fusion in oncogenesis and tumor progression, the agents and approaches used to block cell fusion are not specific for the tumor cells (e.g., blockade of IL-4 receptors impacts many different cells and botulinum toxin digests proteins other than SNARE-type proteins) and the immunodeficiency of the hosts precludes the balancing impact on tumor immunity.
However, if highly specific agents were tested in physiologically normal systems, certain predictions could be proposed. If cell fusion occurs as a consequence of malignancy and/or progression, however, then inhibition of cell fusion should change neither the incidence nor the outcome of cancer. Of course, failure of a cell fusion inhibitor would not prove that cell fusion is a consequence of malignancy, as failure could also reflect the timing of delivery, the dose, or pharmacodynamic or pharmacokinetic limitations of the agent. Failure might also occur if fusion or progression proceeded via several independent pathways.
However, the test of a cell fusion inhibitor could yield another, more interesting result—an increase in the incidence or severity of a malignancy. Thus, while cell fusion has been long considered a potential mechanism of genetic change in malignancy, it has also been found to correct pathogenic changes (see [22,31] for review). Therefore, despite our own research and speculation focusing on pathogenic consequences of cell fusion [3,24,32], we think it prudent to consider whether and how the inhibition of cell fusion might weigh unfavorably in the outcomes of cancer. In doing so, we shall assume that a blocking agent is manifestly effective in vivo and put aside considerations of timing, dose, pharmacokinetics, and pharmacodynamics.
The evolution of multicellularity was associated with, if not dependent on, the evolution of cellular defenses against accumulation of mutations that could spark malignancy [33,34,35]. The evolutionary imperatives included the large number of cell divisions needed to establish the body-plan and sustain tissues, such as intestine and skin, and rapid cell turnover, perhaps in part to limit cumulative exposure to environmental mutagens during longer lifespans. These imperatives presumably selected for stringent and redundant governance of cell proliferation, telomere length, DNA synthesis and repair, and tumor suppression when controls fail [35,36,37]. The specific steps in evolution can be difficult or impossible to prove, but the “selective pressure” can be surmised by comparing risks and adaptations in larger mammals with those of smaller mammals [38]. As Abegglen et al. [36] determined, wild mice, with body mass of ~50 g and lifespan of 4.5 years, and humans should have a vastly lower risk of malignancy than elephants, which reach ~4800 kg body mass and live ~65 years. However, autopsy and necropsy results reveal a similar prevalence of cancers. One explanation sometimes offered is that the evolution of larger body mass and longevity was made possible by evolutionary changes in tumor suppression. Indeed, elephants have ~19 copies of TP53, the product of which arrests cell cycling and induces senescence and apoptosis [39], whereas humans and mice have one copy per haplotype [36,40]. However, given the fecundity and long evolutionary history of rodents, one might as well question why mice do not have more copies of tp53 and as high a prevalence of cancer as humans. The hypothesis we explore is that events, such as fusion, that induce or closely follow upon malignant transformation and progression of cancer could have been appropriated by evolution for defenses against cancer.
The first suggestion that certain genes protect normal cells from malignant transformation emerged from experiments in which normal proliferating cells were deliberately fused with malignant cells [39,41,42]. In 1969, Henry Harris reported that fusion of normal murine fibroblasts with various lines of malignant murine cells led to the formation of stable hybrids that had chromosomal markers of both parental cell lines and did not form tumors in histocompatible mice [31,43]. Reversion of malignant phenotype to normal after fusion of malignant cells with normal cells was soon confirmed using human cells [44]. The absence of tumors in mouse and human hybrids was striking since the malignant parental cells always formed tumors.
As exciting and provocative as the observations of Harris were at that time, it was as apparent then as it is today that malignancy could not be addressed by deliberately fusing normal cells with cancer cells [32]. Rather, Harris drew insights from this model that eventually would transform understanding of malignant transformation and offer clues to potential consequences of the blockade of cell fusion. Thus, Harris also observed that tumor cell-normal cell hybrids occasionally regained the capacity to form tumors. Tumor cell-normal cell hybrids that initially failed to form tumors but reacquired malignancy appeared to have lost chromosomal segments that had originated from the normal parental cells [43]. Harris reasoned that the deleted chromosomal segments included tumor suppressor genes [31]. Harris’s observations thus prompt consideration of the possibility that blocking cell fusion could increase the incidence of de novo malignancy or make existing malignancies worse rather than better.
Our own experience, however, appears to contradict the observations and conclusions one might take from the work of Harris. We conducted experiments designed to determine whether fusion of normal epithelial cells could initiate malignancy [24]. Rat epithelial cells that were manifestly not transformed, had a stable diploid karyotype, and never formed tumors in immunodeficient mice were fused using polyethylene glycol and then cloned. Clones generated from the fused cells frequently exhibited chromosomal instability and aneuploidy, a transformed phenotype, and the capacity to form tumors in immunodeficient mice, consistent with the observations of Harris [43] and others (see [45] for review). Clones that had not fused exhibited none of the features of transformed cells and never initiated tumors in immunodeficient mice. Of note was that aberrant chromosomal numbers or features in a given clone either persisted with little change or reverted toward diploidy, which is to say the propensity for chromosomal damage, translocation, and/or separation in mitosis was transient. We also observed that TP53 in the hybrid clones tested retained wild-type sequence. Since the cells that gave rise to malignancy were cloned after fusion, it is unlikely that wild-type TP53 was generated by reversion, and the results suggest that cell fusion can induce malignancy despite intact tumor suppression pathways. Thus, our findings (and other work) suggest malignant transformation potentially can bypass tumor suppression processes intrinsic to the cell.