

Thapsigargin (Tg), a guaianolide-type sesquiterpene lactone, is abundant in the common Mediterranean weed Thapsia garganica (Apiaceae), known as “deadly carrot” due to its high toxicity to sheep and cattle.
- thapsigargin
- cytotoxin
- anticancer activity
- sarcoplasmic/endoplasmic reticulum Ca2+ ATPase
- unfold protein response
- apoptosis
- prodrug
- prostate-specific antigen
- prostate-specific membrane antigen
- mipsagargin
1. Introduction
Thapsigargin (Tg), a guaianolide-type sesquiterpene lactone, is abundant in the common Mediterranean weed Thapsia garganica (Apiaceae), known as “deadly carrot” due to its high toxicity to sheep and cattle. The skin-irritating properties, as well as the medical use of this plant, were known already in ancient times. The resin from the roots and fruits of T. garganica was used for centuries in folk medicine to treat several diseases, such as pulmonary diseases, female infertility, catarrh, fever, and rheumatism[1][2].
Tg was isolated from T. garganica, along with other structurally-related guaianolides, by Christensen and co-workers in 1978[3][4], whereas the full structure and the absolute configuration of this compound was established in 1985[5].
In terms of chemical structure, Tg consists of three fused rings, formed by annulation of a cycloheptane, cyclopentene, and a γ-lactone (Figure 1). The structural complexity of this carbon skeleton was challenging for organic chemists [2].
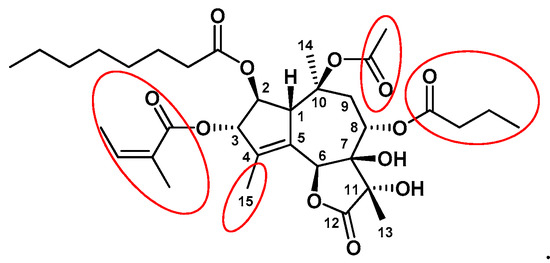
Figure 1. The chemical structure of Tg. Pharmacophoric groups important for biological activity are marked in red circles.
The interesting biological activities of Tg, especially high cytotoxicity, opened perspectives of its use as an anticancer agent, which caused a growing demand for this compound. The low-yield tedious isolation of Tg from T. garganica and laborious and expensive chemical synthesis made the availability of Tg very limited. The solution may lie in the development of production platforms for Tg, including agriculture of T. garganica plants and plant tissue cultures producing Tg, which are already in progress by several companies[1][6], as well as in new, cost-effective synthetic approaches [2].
Over the past 40 years, several excellent reviews describing Tg and its derivatives at different angles have been published [1][2][7][8][9].
2. Molecular Mechanism of Tg Action
2.1. Tg as a SERCA Pump Inhibitor
The sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump, plays a crucial role in regulation of calcium homeostasis which is pivotal for cell signaling and cell survival [10][11]. Impaired SERCA function leads to the elevation of intracellular Ca2+ levels and depletion of Ca2+ in ER stores, triggering ER stress and stress-induced apoptosis[12]. Dysregulation of Ca2+ metabolism may cause cardiovascular and muscular diseases, diabetes and also cancer [10][12][13]. On the other hand, SERCA inhibitors are proposed as novel therapeutic agents in cancer therapy[10][11]. Tg is the best known and most throughly studied representative of SERCA inhibitors [11][14]. Tg blocks SERCA ATPase activity at subnanomolar or low nanomolar concentrations [15][16] and at these concentrations has no influence on either the plasma membrane Ca2+-ATPase or Na+,K+-ATPase[17]. Tg interacts stoichiometrically with the SERCA pump in a calcium-free E2 conformational state, forming an irreversible, catalytically inactive ‘dead-end’ inhibitory complex, which prevents Ca2+ binding and ATPase activation [10][14][18].
2.2. Tg as an Endoplasmic Reticulum Stressor and Unfolded Protein Response (UPR) Inducer
The ER plays a key role in Ca2+ storage and dynamics and is a major site for protein synthesis, folding and maturation of eukaryotic cells[19]. Disturbances of ER functions, leading to protein folding defects and accumulation of unfolded or misfolded proteins in the lumen, results in ER stress[20]. To alleviate this stress, cells activate a network of signaling pathways known as the unfolded protein response (UPR) that, depending on the level of damage, restores protein homeostasis (adaptive UPR) or triggers apoptosis (apoptotic UPR) [20][21][22][23].
Signaling through the UPR increases protein folding, transport and ER-associated protein degradation (ERAD), while inhibiting protein synthesis[23]. The UPR comprises three parallel signaling branches controlled by transmembrane ER sensors: protein kinase R-like ER kinase (PERK), transcription factor-6 (ATF6) and inositol-requiring enzyme 1α (IRE1α) [19][24], as well as the downstream transcription factors: X-box-binding protein 1 (XBP1), activating transcription factor-4 (ATF4) and transcription factor C/EBP homologous protein (CHOP)[22](Figure 2).
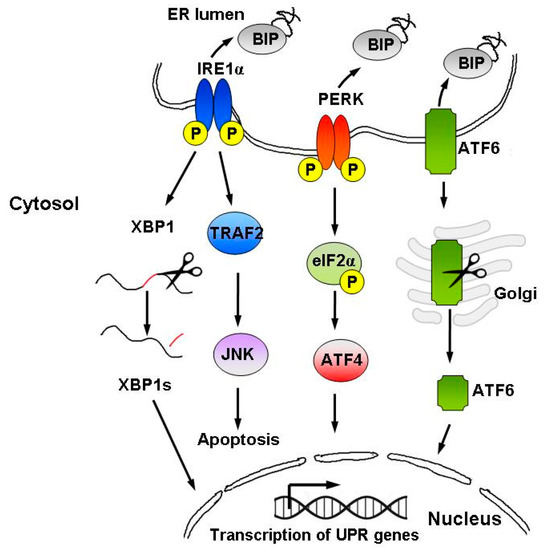
Figure 2. Schematic representation of the UPR signaling pathways. The UPR is activated by accumulation of unfolded proteins in the ER lumen, followed by BiP dissociation from the three ER stress sensors, i.e., IRE1α, PERK, and ATF6. The UPR instigates a transcriptional and translational response to ER stress in order to restore protein homeostasis. However, upon persistent ER stress, pro-apoptotic signaling is induced and the cell undergoes programmed cell death.
When ER stress is persistent and unresolved, UPR triggers specific apoptotic pathways to eliminate severely damaged cells[25][26][27]. Chronic ER stress and defects in UPR signaling may lead to various diseases, including cancer, diabetes, neurodegenerative and autoimmune disorders [28][29]. UPR sits in the center of life-or-death decisions of the cell, activating either pro-survival or pro-apoptotic pathways. Hence, inducing cell death via ER stress and UPR might be a promising therapeutic strategy for killing cancer cells [30].
Tg is known as an ER stressor that initiates UPR-mediated apoptosis [27][31][32] but the contributions of various UPR components involved in cell death initiation remain unclear. In numerous studies on cancer cell lines (Table 1), it has been shown that death receptor 5 (designated DR5, or TRAIL-R2) and caspase-8 play an essential role in Tg-induced apoptosis via UPR (in a response to ER stress) [25][26][31][32][33][34].
Table 1. Cell death mode and key mediators involved in Tg action in various cancer cell lines.
| Mode of Action/Key Proteins | Mediator | Cell Line | Ref |
|---|---|---|---|
| Apoptosis via DR5 | CHOP | Colorectal carcinoma HCT116 Prostate cancer LNCaP, DU145 Ovarian cancer A2780 |
[25] |
| Apoptosis via DR5 and caspase 8 |
Colorectal carcinoma HCT116 Lung cancer SK-MES-1 Myeloma KSM11, RPMI-8226 |
[26] | |
| Apoptosis | Prostate cancer PC3, LNCaP Breast cancer MCF-7 |
[27] | |
| Apoptosis via DR5 and caspase-8 |
LC3B, CHOP, PERK, ATF4, IRE1 | Prostate cancer LNCaP Colorectal carcinoma HCT116 |
[31] |
| Apoptosis via DR5 and caspase 8 |
Colorectal carcinoma HCT116 Cas9 | [32] | |
| Apoptosis via DR5; Inhibition of cell migration, adhesion, and invasion Sensitization to TRAIL-mediated apoptosis via TRAIL-DR5-AMPK signaling |
CHOP, ATF4, PERK, eIF2α; AMPK signaling pathway; ROS |
Esophageal carcinoma EC109, TE12 C109 xenografts in nude mice |
[33] |
| Sensitization to TRAIL-induced apoptosis via DR5 | IRE1α, ATF-6, CHOP | Melanoma Mel-RM, MM200, IgR3, Mel-CV, Me4405, Sk-Mel-28. Mel-FH | [35] |
| Apoptosis via JNK and ERS (JNK/MAPK/ERK signaling); Inhibition of proliferation, migration and invasion |
genes (JNK, ATF6, PERK, LC3B, Bcl-2) proteins (JNK, MAPK, ERK) |
Adrenocortical carcinoma SW-13, NCI-H295R adrenocortical carcinoma xenograft in male BALB/c nude mice |
[36] |
| Apoptosis | SEC24A gene | Near-haploid HAP1 | [37] |
| Apoptosis | XBP1s, ATF4, CHOP, LC3B | Prostate cancer PC3, LNCaP Breast cancer MCF-7 |
[38] |
Linder et al. demonstrated that in human prostate (LNCaP) and colorectal (HCT116) cancer cell lines exposed to Tg, microtubule-associated protein 1A/1B light chain 3B (LC3B), normally associated with autophagy, was required for optimal activation of caspase-8[31]. The authors showed that PERK, and its downstream transcription factors: ATF4 and CHOP, were necessary for Tg-induced cell death but surprisingly they acted in parallel rather than as a linear pathway. The expression of both DR5 and LC3B was controlled by ATF4 and CHOP, whereas PERK was required for cell apoptosis but acted via other pathways[31].
The involvement of CHOP in up-regulation of DR5 and promotion of apoptosis in Tg-treated cells was observed also by other authors[25][33][35]. Chen et al. demonstrated that up-regulation of DR5 in Tg-treated melanoma cells was cooperatively mediated by IRE1α and ATF-6-signaling pathways, along with the transcription factor CHOP[35]. Moreover, they indicated that Tg might be useful in sensitizing melanoma cells to TRAIL-induced apoptosis[35]. Similarly, Ma and colleagues showed that Tg sensitized esophageal squamous cell carcinoma cell line (ESCC) to TRAIL-induced apoptosis via the TRAIL-DR5-AMP activated protein kinase (AMPK) pathway[33]. Detailed studies revealed that Tg-induced ER stress increased CHOP expression, thus up-regulating DR5. TRAIL/DR5 activation induced apoptosis in ESCC cells, which was mediated by oxidative stress via AMPK phosphorylation[33]. Additionally, inducing ER stress, Tg could also directly activate AMPK phosphorylation, which further promoted apoptosis[33]. TRAIL is a member of tumor necrosis factor (TNF) family which, when bound to DR5, can cause apoptosis in a variety of cancer cell types, while sparing normal cells[39]. Therefore, using Tg as an ER stress inducer may be beneficial for improving the efficacy of TRAIL-based anti-cancer agents[33][35].
IRE1α is yet another UPR sensor, whose role in response to ER-stress is complex and not fully elucidated. It controls both cell survival and the decision to execute apoptosis. Activation of IRE1α leads to production of the alternatively spliced isoform of XBP1 (XBP1s), contributing to cell survival during mild, resolvable ER-stress, whereas activation of c-Jun N-terminal kinase (JNK) late in the ER stress response results in cell death[19][40].
The involvement of JNK signaling pathway in Tg-mediated ER stress apoptosis was confirmed in several cancer cell lines and in the in vivo models[31][36][40]. Wu et al. showed that promotion of apoptosis in adrenocortical carcinoma (ACC) cells and suppression of ACC xenograft growth in mice treated with Tg was caused by up-regulation of JNK signaling-related markers[36]. Tg significantly enhanced expression of JNK signaling-related genes (JNK, ATF6, PERK, LC3B, and Bcl-2) and proteins (JNK, MAPK, ERK) and their phosphorylated forms, suggesting activation of JNK/MAPK/ERK signaling pathway in Tg-induced apoptosis in ACC[36]. Recently, it has been suggested that two functionally distinct phases of JNK signaling—An early pro-survival and late pro-apoptotic phase exist in ER stress response and Tg was shown to enhance JNK activity in these both phases[31][40]. Brown et al.[40] demonstrated that the initial phase of JNK activation in response to Tg-induced ER stress depended on both IRE1α and tumor necrosis factor receptor-associated factor 2 (TRAF2), and produced anti-apoptotic signals, protecting cells from executing apoptosis. On the other hand, Linder et al. showed that IRE1-XBP1 was required for the second-phase activation of JNK by Tg, which promoted caspase activation and apoptosis in a cell type-dependent manner[31]. The authors found that Tg induced apoptosis in prostate cancer LNCaP cells, sensitive to pro-death effects of JNK, but not in JNK independent colon cancer HCT116 cells[31].
Recently, Chidawanyika et al.[37] investigated genes engaged in Tg-induced apoptosis. Using a near-haploid cell line HAP1, they identified a novel gene, SEC24A, essential for Tg-induced cytotoxicity in HAP1 cells. They showed that the ability of SEC24A to facilitate ER stress-induced cell death is specific to Tg and that SEC24A is a crucial mediator of Tg-induced UPR and apoptosis.
3. Structure-Activity-Relationship Studies of Tg Analogs
Detailed structure-activity relationship studies were performed to establish the effect of each functional group of Tg on its activity as a SERCA inhibitor[8]. According to the obtained pharmacophoric model, three acyloxy groups (at C-3, C-8, C-10), the methyl group at C-4 and the lactone ring interact with the backbone of SERCA pump via hydrophobic interactions[41][42][43](Figure 1). The hydrophobic chain of the octanoyloxy group at C-2 extends into the lipid phase of a cell membrane forming a weak interaction with ATPase[43]. Detrimental for Tg activity was the inversion of the configuration at C-3 and C-8[8][41], while long, flexible chains of acyloxy groups at C-8 did not reduce Tg’s inhibitory potency against SERCA. Therefore, modifications at C-8 became of particular interest in the design of new analogs. Replacing the butanoiloxy group at C-8 by a 12-aminododecanoiloxy moiety gave 8-(12-aminododecanoyloxy)thapsigargin, which was further modified by the attachment of amino acid residues to form Leu-12ADT (Figure 3) and βAsp-12ADT (Figure 4)[27][44][45].
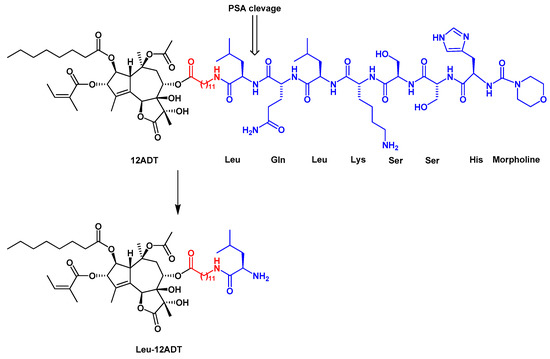
Figure 3. The structure of the G115 prodrug and the product of its PSA cleavage, Leu-12ADT. The linker is marked in red.
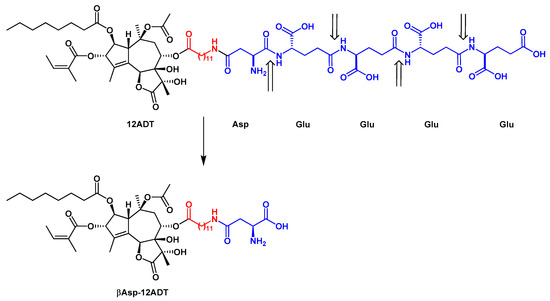
Figure 4. The structure of G202 prodrug and the major product of its PSMA cleavage, βAsp-12ADT. The linker is marked in red. The PSMA cleavage sites are indicated by arrows.
4. Development of Tg-Based Prodrugs
Through the inhibition of SERCA pump, Tg induces cancer cell apoptosis in a proliferation-independent manner in both proliferative and quiescent phases of the cell cycle[44][46]. However, the presence of SERCA pump in almost all kind of cells and its essential role for cell survival, makes Tg a general cell toxin[44][46]. This high toxicity excludes Tg as a drug candidate. To overcome this limitation and direct the cytotoxicity of Tg towards cancer cells, sparing normal tissue, the Tg-based pro-drug strategy has been developed[1][9][44][45][46].The Tg-based prodrug strategy utilizes the proteolytic enzymes that are preferentially expressed on the surface of cancer cells or are secreted by cells of solid tumors. The conjugation of Tg with substrates for such enzymes masks cytotoxic activity of Tg, producing prodrugs that can be cleaved by a tissue-specific proteases expressed only in cancer cells[44][45][46]. By coupling Tg derivatives to peptide carriers, which are substrates either for prostate-specific antigen (PSA) or prostate specific membrane antigen (PSMA), protease-activated prodrugs, selectively affecting prostate cancer cells, were created[44][45][46]. The successful examples of such approach are two prodrugs, G115[44][46], designed for the treatment of prostate cancer, and G202, selectively targeting the neovascular tissue in blood vessels of various cancer types, including prostate, breast, and bladder cancers[45][47][48]. In vivo experiments on a variety of human cancer xenografts in mice reveled that G202 significantly inhibited progression of prostate, breast and bladder cancers, while being minimally toxic for the host animals[45]. After compelling preclinical results (low toxicity in monkeys), G202 has entered clinical trials, under the name mipsagargin. Phase I clinical trials demonstrated that mipsagargin was well tolerated and had favorable pharmacokinetic profile in patients with advanced solid tumors, resistant to standard therapy[47]. Phase II clinical trials on mipsagargin are summarized in Table 2 (http://clinicaltrials.gov; November 2020).
Table 2. Phase II clinical trials of mipsagargin.
|
Cancer Type |
Status |
Gov Identifier |
|
Glioblastoma multiforme |
Completed |
NCT02067156 |
|
Prostate cancer |
Withdrawn |
NCT01734681 |
|
Advanced Adult Hepatocellular Carcinoma |
Completed |
NCT01777594 |
|
Glioblastoma |
Withdrawn prior to enrolment |
NCT02876003 |
|
Clear Cell Renal Cell Carcinoma |
Completed |
NCT02607553 |
|
Prostatic Neoplasms |
Completed |
NCT02381236 |
|
Hepatocellular Carcinoma |
No longer available |
NCT02082691 |
Antineoplastic activity of Tg-based prodrugs has been widely described in many types of solid tumors. On the other hand, Roti et al. demonstrated antileukemic activity of Tg in human T cell acute lymphoblastic leukemia (T-ALL)[49]. Moreover, by conjugation of the folic acid to an alcohol derivative of Tg via a cleavable ester bond, Roti et al. designed a Tg-folate prodrug JQ-FT. This molecule was recognized by folate receptors (FRs) on the plasma membrane and delivered into leukemia cells. Folate-derived Tg delivery strategy is an interesting approach to enhance the therapeutic window of Tg, providing dual selectivity: leukemia over normal cells and NOTCH1 mutated over wild-type receptors. However, further optimization and potency enhancement is required for clinical application of this molecule[50].
5. Conclusions and Future Perspectives
Natural products isolated from plants are an important source of chemotherapeutics, in particular, against cancer. However, in general, natural substances cannot be used directly as drugs, either because they have a narrow therapeutic window or because they possess undesired pharmacokinetic properties, such as poor absorption, low solubility, and/or fast metabolism. Therefore, optimization of the properties of natural phytochemicals is required. The solution lies in structural modifications of natural compounds or synthesis of their analogs with improved pharmacological properties.
Tg is a potent cytotoxin isolated from T. garganica over forty years ago. Tg induces apoptosis in a proliferation-independent manner by inhibiting the SERCA pump and emptying the ER Ca2+ stores. A barrier preventing the direct usage of Tg as an anticancer agent is its lack of selectivity, since Tg kills not only cancer but also normal cells. The unique properties of cancer cells were used to develop prodrugs that can transport Tg directly to cancer sites. G115 and G202 are examples of prodrugs obtained by conjugation of Tg to substrates of proteolytic enzymes, that are present only in cancer tissue. JQ-FT is an antileukemic prodrug developed by conjugation of folic acid and Tg derivative. Both described prodrug strategies represent an efficient approach to overcome Tg general cytotoxicity, and are in line with recent directions in targeted cancer therapy.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22010004
References
- Andersen, T.B.; López, C.Q.; Manczak, T.; Martinez, K.; Simonsen, H.T. Thapsigargin—From Thapsia L. to mipsagargin. Molecules 2015, 20, 6113–6127.
- Dey, S.; Bajaj, S.O. Promising anticancer drug thapsigargin: A perspective toward the total synthesis. Synth. Commun. 2018, 48, 1–13.
- Rasmussen, U.; Christensen, S.B.; Sandberg, F. Thapsigargine and thapsigargicine, two new histamine liberators from Thapsia garganica L. Acta Pharm. Suec. 1978, 15, 133–140.
- Smitt, U.W.; Christensen, S.B. Nortrilobolide, a new potent guaianolide secretagogue from Thapsia garganica. Planta Med. 1991, 57, 196–197.
- Christensen, S.B.; Norup, E. Absolute configurations of the histamine liberating sesquiterpene lactones thapsigargin and trilobolide. Tetrahedron Lett. 1985, 26, 107–110.
- Makunga, N.P.; Jäger, A.K.; van Staden, J. Improved in vitro rooting and hyperhydricity in regenerating tissues of Thapsia garganica L. Plant Cell Tiss. Organ Cult. 2006, 86, 77–86.
- Christensen, S.B.; Skytte, D.M.; Denmeade, S.R.; Dionne, C.; Møller, J.V.; Nissen, P.; Isaacs, J.T. A Trojan horse in drug development: Targeting of thapsigargins towards prostate cancer cells. Anticancer Agents Med. Chem. 2009, 9, 276–294.
- Doan, N.T.; Christensen, S.B. Thapsigargin, Origin, Chemistry, Structure-Activity Relationships and Prodrug Development. Curr. Pharm. Des. 2015, 21, 5501–5517.
- Doan, N.T.Q.; Paulsen, E.S.; Sehgal, P.; Møller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Dionne, C.A.; Christensen, S.B. Targeting thapsigargin towards tumors. Steroids 2015, 97, 2–7.
- Tadini-Buoninsegni, F.; Smeazzetto, S.; Gualdani, R.; Moncelli, M.R. Drug Interactions With the Ca2+-ATPase From Sarco(Endo)Plasmic Reticulum (SERCA). Front. Mol. Biosci. 2018, 5, 36.
- Peterková, L.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Sarco/Endoplasmic Reticulum Calcium ATPase Inhibitors: Beyond Anticancer Perspective, J. Med. Chem. 2020, 63, 1937–1963.
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61.
- Rahate, K.; Bhatt, L.K.; Prabhavalkar, K.S. SERCA stimulation: A potential approach in therapeutics. Chem Biol Drug Des. 2020, 95, 5–15.
- Michelangeli, F.; East, J.M. A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Trans. 2011, 39, 789–797.
- Wootton, L.L.; Michelangeli, F. The effects of the phenylalanine 256 to valine mutation on the sensitivity of sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) Ca2+ pump isoforms 1, 2, and 3 to thapsigargin and other inhibitors. J. Biol. Chem. 2006, 281, 6970–6976.
- Sagara, Y.; Inesi, G. Inhibition of the sarcoplasmic reticulum Ca2+ transport ATPase by thapsigargin at subnanomolar concentrations. J. Biol. Chem. 1991, 266, 13503–13506.
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991, 266, 17067–17071.
- Sagara, Y.; Wade, J.B.; Inesi, G. A conformational mechanism for formation of a dead-end complex by the sarcoplasmic reticulum ATPase with thapsigargin. J. Biol. Chem. 1992, 267, 1286–1292.
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11.
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838.
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell. 2018, 69, 169–181.
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652.
- Qi, L.; Tsai, B.; Arvan, P. New Insights into the Physiological Role of Endoplasmic Reticulum-Associated Degradation. Trends Cell Biol. 2017, 27, 430–440.
- Karagöz, G.E.; Acosta-Alvear, D.; Walter, P. The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2019, 11, a033886.
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502.
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101.
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673.
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194.
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335.
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597.
- Lindner, P.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal. 2020, 18, 12–23.
- Lam, M.; Lawrence, D.A.; Ashkenazi, A.; Walter, P. Confirming a critical role for death receptor 5 and caspase-8 in apoptosis induction by endoplasmic reticulum stress. Cell Death Differ. 2018, 25, 1530–1531.
- Ma, Z.; Fan, C.; Yang, Y.; Di, S.; Hu, W.; Li, T.; Zhu, Y.; Han, J.; Xin, Z.; Wu, G.; et al. Thapsigargin sensitizes human esophageal cancer to TRAIL-induced apoptosis via AMPK activation. Sci. Rep. 2016, 6, 35196.
- Muñoz-Pinedo, C.; López-Rivas, A. A role for caspase-8 and TRAIL-R2/DR5 in ER-stress-induced apoptosis. Cell Death Differ. 2018, 25, 226.
- Chen, L.H.; Jiang, C.C.; Kiejda, K.A.; Wang, Y.F.; Thorne, R.F.; Zhang, X.D.; Hersey, P. Thapsigargin sensitizes human melanoma cells to TRAIL-induced apoptosis by up-regulation of TRAIL-R2 through the unfolded protein response. Carcinogenesis 2007, 28, 2328–2336.
- Wu, L.; Huang, X.; Kuang, Y.; Xing, Z.; Deng, X.; Luo, Z. Thapsigargin induces apoptosis in adrenocortical carcinoma by activating endoplasmic reticulum stress and the JNK signaling pathway: An in vitro and in vivo study. Drug Des. Dev. Ther. 2019, 13, 2787–2798.
- Chidawanyika, T.; Sergison, E.; Cole, M.; Mark, K.; Supattapone, S. SEC24A identified as an essential mediator of thapsigargin-induced cell death in a genome-wide CRISPR/Cas9 screen. Cell Death Discov. 2018, 4, 115.
- Szalai, P.; Parys, J.B.; Bultynck, G.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Nonlinear relationship between ER Ca2+ depletion versus induction of the unfolded protein response, autophagy inhibition, and cell death. Cell Calcium 2018, 76, 48–61.
- Wong, S.H.M.; Kong, W.Y.; Fang, C.M.; Loh, H.S.; Chuah, L.H.; Abdullah, S.; Ngai, S.C. The TRAIL to cancer therapy: Hindrances and potential solutions. Crit. Rev. Oncol. Hematol. 2019, 143, 81–94.
- Brown, M.; Strudwick, N.; Suwara, M.; Sutcliffe, L.K.; Mihai, A.D.; Ali, A.A.; Watson, J.N.; Schröder, M. An initial phase of JNK activation inhibits cell death early in the endoplasmic reticulum stress response. J. Cell Sci. 2016, 129, 2317–2328.
- Søhoel, H.; Jensen, A.M.; Møller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Olsen, C.E.; Christensen, S.B. Natural products as starting materials for development of second-generation SERCA inhibitors targeted towards prostate cancer cells. Bioorg. Med. Chem. 2006, 14, 2810–2815.
- Skytte, D.M.; Møller, J.V.; Liu, H.; Nielsen, H.Ø.; Svenningsen, L.E.; Jensen, C.M.; Olsen, C.E.; Christensen, S.B. Elucidation of the topography of the thapsigargin binding site in the sarco-endoplasmic calcium ATPase. Bioorg. Med. Chem. 2010, 18, 5634–5646.
- Winther, A.M.; Liu, H.; Sonntag, Y.; Olesen, C.; le Maire, M.; Soehoel, H.; Olsen, C.E.; Christensen, S.B.; Nissen, P.; Møller, J.V. Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. J. Biol. Chem. 2010, 285, 28883–28892.
- Denmeade, S.R.; Jakobsen, C.M.; Janssen, S.; Khan, S.R.; Garrett, E.S.; Lilja, H.; Christensen, S.B.; Isaacs, J.T. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J. Natl. Cancer Inst. 2003, 95, 990–1000.
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; Demarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140ra86.
- Denmeade, S.R.; Isaacs, J.T. The SERCA pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol. Ther. 2005, 4, 14–22.
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994.
- Mahalingam, D.; Peguero, J.; Cen, P.; Arora, S.P.; Sarantopoulos, J.; Rowe, J.; Allgood, V.; Tubb, B.; Campos, L. A Phase II, Multicenter, Single-Arm Study of Mipsagargin (G-202) as a Second-Line Therapy Following Sorafenib for Adult Patients with Progressive Advanced Hepatocellular Carcinoma. Cancers 2019, 11, 833.
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell. 2013, 23, 390–405.
- Roti, G.; Qi, J.; Kitara, S.; Sanchez-Martin, M.; Saur Conway, A.; Varca, A.C.; Su, A.; Wu, L.; Kung, A.L.; Ferrando, A.A.; et al. Leukemia-specific delivery of mutant NOTCH1 targeted therapy. J. Exp. Med. 2018, 215, 197–216.
