1. Voltage-Dependent Ca2+ Channels
Extracellular Ca
2+ entry is primordial in Ca
2+ signaling, and its influx is primarily driven by an electrochemical gradient. In ASM, both L- type VDCCs (Long Lasting Currents voltage-dependent Ca
2+ channels) and T-type VDCCs (Transient Currents voltage-dependent Ca
2+ channels) are expressed, but L-VDCC is the predominant type in human ASM and various other species [
24,
25,
26,
27,
28,
29,
30,
31,
32,
33]. In this sense, the preincubation with E2 at physiological levels can inhibit L-VDCCs non-genomically (
Figure 3) in a concentration-dependent manner in human ASM (hASM) stimulated with histamine, with a more pronounced effect when using an ERα-selective agonist, which was not observed when using an ERβ selective agonist (
Table 1) [
20]. This could be partially due to the localization of the receptors since ERβ is minimally present in the plasma membrane, where non-genomic effects could take place [
20]. This effect seems to have a biphasic response since at supraphysiological levels, E2 also inhibits L-VDCCs non-genomically in guinea pig ASM, as demonstrated through electrophysiological studies (
Table 1) [
34]. Both estrogen receptors seem to have different signaling pathways. They serve distinct purposes, both in physiological and pathological conditions. For instance, in human ASM cells from an asthmatic, an increase in the expression of the different variants of both receptors at different degrees has been reported, although their role in asthma pathophysiology remains to be elucidated [
21]. Furthermore, chronic exposure (24 h, genomic effect) to estrogens in hASM cells from asthmatics and non-asthmatics seems to have opposing effects on [Ca
2+]i. This phenomenon seems to depend on the type of receptor activated. An ERα-specific agonist ((R,R)-THC) augmented the [Ca
2+]i response induced by histamine in both asthmatic and non-asthmatic hASM cells. Meanwhile, an ERβ-specific agonist (DPN), decreased the [Ca
2+]i response induced by histamine in asthmatic and non-asthmatic hASM cells. The effect observed on the [Ca
2+]i response with activation of ERβ signaling appears to implicate the inhibition of L-VDCC (
Figure 3) (
Table 1) [
35]. Interestingly, E2, (as a non-selective ER agonist) at physiological concentrations did not show significant changes in the [Ca
2+]i response to different agonists in ASM [
35]. Unfortunately, the genomic effects of estrogen on L-VDCC expression, have not been explored in ASM cells yet. In comparison, ovariectomy (OVX) induced an increase in the channels’ expression in rat aorta. Interestingly, treatment with E2 downregulated the channels’ mRNA expression, and treatment with E2 and tamoxifen (an ER blocker) had a similar effect as E2 alone [
36].
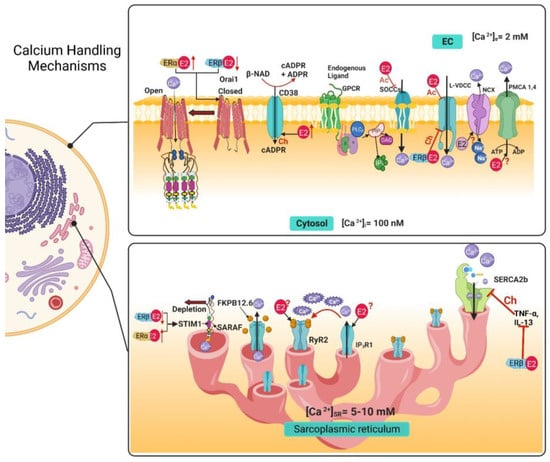
Figure 3. Calcium handling mechanisms in airway smooth muscle and their modulation by estrogens. E2 through non-genomic effects inhibits SOCCs, and the chronic exposure to an ERβ-specific agonist decreases the expression of STIM1 and Orai1, and an ERα-specific agonist increases the expression of STIM1 and Orai1.The genomic effect of E2 increases the expression of CD38, although the non-genomic effect is still unknown. E2 acutely inhibits the LVDCC and the chronic exposure, via ERβ pathway, inhibits this channel. In the sarcoplasmic reticulum, the chronic exposure with an ERβ-specific agonist inhibits the increase in SERCA expression caused by exposure to TNF-α and IL-13. The genomic and non-genomic effects of E2 over RyR2, IP3R1, NCX and PMCA are still not reported. EC, extracellular; Ac, Acute; Ch, Chronic; IP3, inositol 1,4,5-trisphosphate; [Ca2+]i, intracellular calcium concentration; [Ca2+]e, extracellular calcium concentration; [Ca2+]SR, sarcoplasmic reticulum calcium concentration; E2, 17β estradiol; ERα, estrogen receptor α; ERβ, estrogen receptor β; L-VDCC, L-type voltage dependent Ca2+ channel; SOCC, store-operated Ca2+ channel, NCX, Na+/Ca2+ Exchanger; PMCA, plasma membrane Ca2+ ATPase; IP3R, inositol 1,4,5-trisphosphate receptor; RyR, Ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+ ATPase; FKBP-12.6, 12.6 kDa FK506-binding protein; Orai1; STIM1, stromal interacting molecule; SARAF, Store-operated Ca2+ entry-associated regulatory factor.
Table 1. Summary of the effects of estrogens on the calcium handling mechanisms in the airway smooth muscle.
| |
Acute |
Chronic |
| Calcium Handling Mechanisms |
Pathway |
Effect |
Pathway |
Effect |
| Voltage-dependent Ca2+ channels (VDCCs) |
ERα |
Inhibition [20,34] |
ERβ |
Inhibition [35] |
| Store-Operated Calcium Channels (SOCCs) |
ERα |
Inhibition via STIM1 phosphorylation [20,50] |
ERβ |
Downregulated STIM1 and Orai1 expression [49] |
| ERα |
Upregulated STIM1 and Orai1 expression [49] |
| Ryanodine Receptor (RyR) |
Unknown |
Unknown |
ERs |
Upregulates CD38 expression [64] |
| IP3 Receptor (IP3R) |
Unknown |
Unknown |
Unknown |
Unknown |
| Na+/Ca2+ Exchanger (NCX) |
No effect |
No effect [20] |
Unknown |
Unknown |
| Plasma Membrane Ca2+ ATPase (PMCA) |
Unknown |
Unknown |
Unknown |
Unknown |
| Sarcoplasmic Reticulum Ca2+ ATPase (SERCA) |
No effect |
No effect |
ERβ |
Upregulates SERCA2 expression [35] |
2. Store-Operated Calcium Channels
Another group of calcium channels that regulates Ca
2+ influx to the cytosol is SOCCs, considered non-selective cation channels. Its activity depends on the SR Ca
2+depletion and its goal is to contribute to the refilling of internal Ca
2+ stores [
37]. To carry out their function following SR depletion, these channels must assemble into a complex formed by two proteins: Orai1 (calcium release-activated calcium channel protein 1) and STIM1 (stromal interacting molecule) [
37]. When at rest, STIM1 is bound to Ca
2+. When Ca
2+ levels begin to diminish in the SR, STIM1 disassociates from Ca
2+, and this change causes STIM1 molecules to cluster and translocate to a region in proximity to the plasma membrane [
37,
38,
39]. Orai1 is a transmembrane protein located in the plasma membrane. In basal conditions, this protein is a dimer, but when STIM1 clusters are formed, they interact with Orai1 and enable them to form tetramers, forming selective Ca
2+ pores that allow Ca
2+ influx [
37,
39]. This mechanism is negatively regulated by the SR transmembrane protein SARAF (store-operated Ca
2+ entry-associated regulatory factor). When SARAF interacts with STIM1, it prevents spontaneous activation or the interaction between STIM1-Orai1 [
37,
40,
41]. On the other hand, transient receptor potential canonical (TRPC) channels also play an important role in Ca
2+ homeostasis, and it is known that the TRPC3 isoform prevails in ASM cells [
24]. Recently, the STIM1-Orai1 complex was found to interact with TRPC channels required for their activation [
42,
43]. Other members of the TRP channel family are vanilloid (TRPV) receptors, ankyrin (TRPA) and melastatin (TRPM), of which TRPV1 and TRPV4 have been identified in ASM cells (
Figure 3) [
24,
44,
45,
46,
47].
Given the importance of SOCCs in Ca
2+ homeostasis, the effects that E2 could have on their regulation is of increasing interest. Townsend et al. determined that the decrease in Ca
2+ induced by the acute exposure to physiological concentrations of E2 observed in the Ca
2+ response induced by histamine in hASM cells (
Table 1) [
20], was in part due to inhibition of SOCCs observed as a diminished SR refilling (
Figure 3) [
48]. On the other hand, the chronic effects of the estrogen receptor signaling on SOCCs modulation have also been explored in healthy and asthmatic hASM cells. The chronic exposure (24 h) to an ERβ-selective agonist (WAY-200070) downregulated the expression of STIM1 and Orai1 measured through Western blot (
Figure 3), and consequently produced a decrease in [Ca
2+]i. This effect was observed in hASM from non-asthmatics, but was more pronounced in asthmatics [
49]. When exposed to an ERα-selective agonist (propyl pyrazole triol at 10 nM) for 24 h, an opposite effect was observed. SOCCs Ca
2+ influx was increased, and the expression of STIM1 and Orai1 was also increased (
Figure 3) (
Table 1) [
49].
Cigarette smoke (CS) is a common risk factor associated with many airway diseases, and asthmatic women exposed to CS tend to have a worse asthmatic response; therefore, the effects of CS and E2 on ASM Ca
2+ regulation were explored [
50]. As a result, it was defined that, in hASM cells, a 24 h exposure to CS extract induces a significant increase in the Ca
2+ response to histamine. On the other hand, acute exposure to nanomolar E2 concentrations inhibits the Ca
2+ response to histamine partially via inhibition of SOCCs [
20]. When exposed to CS extract, this E2 effect was blunted [
50]. Chronic exposure to CS extract (24h exposure to 1% or 2% CS) also seems to increase the expression of ERα and ERβ, leading to investigate if a differential ER regulation was present. As observed before, the acute effect on Ca
2+ was present when using an ERα-specific agonist but not an ERβ-selective agonist [
20]. This effect was absent when the cells were previously exposed to CS extract [
50]. Therefore, CS extract enhances [Ca
2+]i through the dysregulation of ER signaling, and blunts the acute reduction in [Ca
2+]i and subsequent force generation resultant from ERα activation, more so in asthmatic patients than in healthy subjects. E2 also non-genomically inhibited STIM1 phosphorylation, while pre-exposure to CS extract for 24 h abolished this E2 effect [
50].
Studies in other cellular types point out the physiological implications of E2-mediated regulation of SOCCs. In airway epithelial cells, E2 (10 nM) acute (15 min) exposure can reduce STIM1 phosphorylation, preventing the formation of STIM1 clusters from interacting with and activating Orai1, decreasing SOCCs activity. This decrease in SOCCs influx also affects Ca
2+ activated Cl
− channels colocalized with Orai1, impacting mucus hydration [
51]. In mouse embryonic stem cells (mESCs), the treatment with physiological concentrations of E2 (1 pM and 1nM) during 24 h enhanced cellular proliferation in a concentration-dependent manner. The effect was through SOCCs activity and could be reverted by SOCCs blockers (2-APB 0.3 µM) [
52]. As already mentioned, Ca
2+ homeostasis disruption can lead to pathologies. Such is the case in various cancer types, where dysfunction of Ca
2+ homeostasis has been implicated. Epithelial ovarian cancer (EOC) is closely tied to E2 regulation, and the effects that E2 could have over Orai1 and the different pathological processes in EOC have been explored [
53]. After 12 h exposure with E2 (at a micromolar range), Orai1 expression increased in SK-OV-3 cells (an EOC cell line), with the most significant effect observed at 1 µM, leading to an increase in [Ca
2+]i. E2, through Orai1, positively regulated cellular and migration (by CDK6 and MMP-1 pathways, respectively), and suppressed cellular proliferation apoptosis through caspase3 expression regulation [
53]. In another study in EOC cells, the chronic exposure to E2 (10 nM–1 µM for 24, 48, and 72 h) increased the mRNA levels and protein expression of TRPC3, and via TRPC3 increased cellular proliferation and migration [
54]. E2 upregulates TRPV1 expression, participating in pain induction, endometriosis and bone resorption. TRPV1 mRNA levels have been shown to be decreased by E2. Through GPR30, E2 modulates TRPV1 phosphorylation to participate in pain sensitization. Through non-genomic effects, E2 has been shown to both potentiate and decrease capsaicin-evoked currents of TRPV1 [
55]. Yang et al., found that TRPV6 expression was increased in human endometrium after E2 treatment [
56]. Similarly, in mouse uterine tissues, TRPV5 and TRPV6 were upregulated with E2 treatment; bisphenol A (BPA) that has estrogenic effects, also significantly increased TRPV5 and TRPV6 expression but not to the degree of E2 [
57]. Upregulation of TRPA1 by E2 participates in the pathophysiology of endometriosis. Furthermore, through non-genomic effects, E2 increments TRPA1 activation in glucose-induced insulin secretion [
55].
Through these studies, we can observe the diverse interactions that estrogens can have over SOCCs modulation and the repercussions in different pathological states.
3. Ryanodine Receptor
In the SR, one of the mechanisms in charge of the Ca
2+ efflux to the cytosol is the RyR. In mammals, three isoforms are found -1, -2, and -3; in mouse ASM, RyR1, and RyR2 are the predominant isoforms, with minimal expression of RyR3 [
23,
24,
58]. It has been found that he endogenous modulation of RyR activity in ASM cells is through the CD38/Cyclic ADP-ribose signaling pathway, regulating [Ca
2+]i. The membrane-bound protein CD38 synthesizes or degrades cADPR, which functions as a ligand for the protein FKBP-12.6 (12.6 kDa FK506-binding protein). In turn, FKBP112.6 binds to the RyR, providing stabilization and reducing the opening probability of the channel. When cADPR binds to the regulatory protein, this causes a conformational change in the RyR, activating Ca
2+ release to the cytosol [
59,
60,
61]. Alterations in this signaling pathway, such as the upregulation of CD38 induced by TNF-α, IL-1β, and IFN-γ, can lead to an asthmatic phenotype of the ASM cells, characterized by airway hyperresponsiveness (AHR) that might develop when an already enhanced [Ca
2+]i concentration is increased further after Gq protein coupled receptor (GPCR) activation [
59,
61]. Until now, no non-genomic effects of estrogens over RyRs in ASM cells have been reported. However, in rat cardiomyocytes, bisphenol A (BPA) and bisphenol S (BPS) (estrogenic endocrine-disrupting chemicals) have shown acute effects over RyR activity [
62,
63]. At nanomolar concentrations, both substances significantly altered characteristic RyR-mediated SR Ca
2+ sparks by transiently and rapidly (30s–5 min) increasing the phosphorylation of RyR at the serine 2808 site through protein kinase A(PKA) activity, as well as phospholamban (PLB, a protein that binds to SERCA and regulates its activity) by Ca
2+/CaM-dependent protein kinase II (CAMKII). This effect was completely abolished when a selective-ERβ blocker was used (PHTPP), but not with a selective-ERα blocker (MPP), indicating that this effect is dependent on the ERβ signaling pathway [
62,
63].
In mouse ASM cells, long term exposure (24–48 h) to E2 (at physiological levels) was shown to upregulate the expression of CD38 (
Figure 3) (
Table 1) [
64]. As mentioned before, CD38 is a key component in the mechanisms in charge of Ca
2+ homeostasis by regulating the activity of RyRs. The genomic effect that estrogens could have directly on RyR expression in ASM cells has yet to be explored. In other models, estrogens have shown to impact RyR expression. In uterine arteries from pregnant sheep (a period of higher estrogenic levels), all three isoforms of RyR were upregulated, causing an increase in Ca
2+ sparks. Additionally, in uterine arteries from nonpregnant sheep treated ex vivo with estrogen and progesterone mimicking pregnancy conditions, a similar upregulation of RyR was observed [
65]. In female rat cardiomyocytes, RyR2 is expressed at higher levels than in male rats [
66,
67,
68]. In contrast, other reports state that in female cardiomyocytes, RyR2 phosphorylation by CaMKII/PKA is reduced, causing lower Ca
2+ sparks [
66,
69,
70]. Another possible mechanism of modulation through the ER signaling pathway is by direct protein–protein interaction. Recently, it was discovered that ERβ has an atypical non-genomic effect over the RyR, in the neuronal cell line HT-22. In these cells, RyR2 and ERβ have varying levels of co-localization, and in electrophysiological studies using RyRs from mouse brain incorporated into artificial lipid bilayers, the application of unliganded (E2-free) ERβ1 monomers caused a significant increase in single-channel currents under basal [Ca
2+]i of 100 nM. This effect caused by the addition of ERβ1 could indicate a synergic interaction with Ca
2+ and RyR that increases the open probability of the channel and could potentiate RyR activity of Ca
2+-induced Ca
2+-release [
71]. The modulatory activity that the complex E2-ERβ1, or the other isoforms of the ERβ and ERα, could have on the RyR remains to be determined. It might be possible that RyR-ERs interact in the ASM. This probability warrants further and more precise studies.
4. IP3 Receptor
The other mechanism that releases Ca
2+ from the SR is the IP
3R, activated by its agonist IP
3 (inositol 1,4,5-trisphosphate) [
23,
24,
37]. As extensively known, when an agonist binds to a membrane GPCRs, phospholipase Cβ (PLCβ) is activated and hydrolyzes the lipid phosphatidylinositol 4,5-biphosphate, generating IP
3 and diacylglycerol (DAG) [
23,
24,
37]. In mammals, three isoforms of the IP
3R are expressed (IP
3R1, -2, and -3); all isoforms have been identified in ASM (
Figure 3) [
24,
72,
73,
74,
75]. Afterward, IP
3 binds to the IP
3R in the SR, generating the release of Ca
2+ from these internal stores. This pathway has usually been implicated in the agonist-induced contraction of the ASM. However, IP
3R participation in the bronchodilation mechanism induced by the TAS2R pathway (Type 2 taste receptor) has also been proposed [
76,
77].
The genomic and non-genomic effects that estrogens could have on the IP
3 signaling pathway in ASM cells remain to be explored, but estrogen modulation in other cell types has been reported (
Figure 3). E2 (1 nM) increased IP
3 production after 6 h of exposure (non-genomic effect) in rat oviduct smooth muscle cells, a phenomenon mediated through an increase in PLC activity [
78]. In HEPG2 cells (human hepatoma cell line), the addition of E2 (nanomolar range) induced a rapid increase in IP
3 production [
79], which was also observed in female rat chondrocytes [
80]. Treatment of rat osteoblasts with E2 (100 pM) caused rapid transient increases in [Ca
2+]i via the PLCβ-IP
3 pathway [
81]. E2 treatment has been shown to cause rapid increases in [Ca
2+]i through an ER interaction with type 1a metabotropic glutamate receptors (mGluR1a), activating the PLCβ-IP
3 pathway in female rat astrocytes [
82]. Among the genomic effects, it has been reported that IP
3R1 expression is suppressed after 48h exposure to E2 (10 nM) in human g-292 osteosarcoma cells and rat osteoblasts [
83]. In rat choroidal plexus epithelial cells, E2 (nanomolar range) downregulated the expression of receptors TAS2R109 and -144, as well as PLCβ2, resulting in a decrease in [Ca
2+]i in response to TAS2R agonists [
77,
84].
5. Na+/Ca2+ Exchanger
During the membrane depolarization phase in ASM, an accumulation of Na
+ underneath the plasma membrane takes place, influencing the Ca
2+ homeostasis through the generation of local concentration gradients. The consequent modulation of the [Ca
2+]i through the Na
+ gradients results in various physiological processes, depending on the magnitude, time and region, including contraction, proliferation, protein synthesis and apoptosis, among others [
85].
The NCX serves as one of the [Ca
2+]i buffer mechanisms by extruding Ca
2+ from the cytosol to the extracellular space [
23,
24]. The NCX carries three Na
+ ions into the cytosol while extruding one Ca
2+; three isoforms, NCX1, -2, and -3 are known, and the most prominent isoform in ASM is the variant NCX1.3 [
24,
86,
87,
88,
89]. The participation of NCX in Ca
2+ homeostasis in ASM seems to be minor (
Figure 3) [
24,
90], and apparently its reverse mode (NCX
rev) has higher importance in ASM physiology. Interestingly, NCX
rev introduces Ca
2+ and extrudes Na
+ [
24]. NCX
rev plays a preponderant role in agonist-stimulated ASM, as for instance, an inhibitor of NCX
rev (KB-R7943, 10 µM) attenuated the [Ca
2+]i increase and contraction induced by carbachol (CCh 100 µM) [
91]. The Ca
2+ influx caused by the removal of Na
+ in hASM and mouse ASM was blocked by KB-R7943 [
91].
Seemingly, NCX
rev also has an important function during oscillatory contractions in mouse ASM induced by potassium channel blockade with tetraethylammonium chloride (TEA). In this experiment, two pattern changes of [Ca
2+]i were induced. One was a high-frequency oscillation, and the other a low-frequency rhythmic oscillation. Both types of Ca
2+ changes participate in triggering ASM contraction, and they might participate in other physiological processes in the ASM. Remarkably, these oscillations augment [Ca
2+]i, an increase that activates NCX, initiating the relaxation phase by extruding Ca
2+ [
92]. Alterations of the NCX also participate in pathological conditions; TNF-α or IL-13 treatment of hASM cells upregulated the expression of NCX1, and the treatment with KB-R7943 abolished methacholine induced AHR in an allergic mouse model [
91,
93]. This was also observed in a chronic allergen-induced AHR murine model, where NCX1 was upregulated and had higher NCX
rev activity [
94].
The effects of E2 on NCX modulation have not been as extensively investigated as other mechanisms. The acute exposure to E2 in hASM cells did not have a significant effect on NCX (
Table 1) [
20], and the effects of a chronic exposure to E2 on NCX in ASM cells has not been explored yet (
Figure 3). Likewise, the participation of NCX in other tissues’ physiology is inconclusive. The chronic treatment with E2 (3 days) in prepubescent female rats resulted in downregulation of NCX1 expression in the esophagus [
95]. In female rat cardiomyocytes, OVX (ovariectomy) caused downregulation of NCX expression that was reversed by E2 treatment [
66,
96]. Similarly, in female rabbit cardiomyocytes, NCX expression was greater than in males [
66,
97,
98], and cardiomyocytes incubated with E2 1 nM during 24 h presented a 50% increase in NCX1 protein expression and I
NCX density mediated by ERs [
98]. Contrary to these findings, OVX or E2 replacement therapy did not alter NCX expression in rat cardiomyocytes. However, NCX activity was significantly increased after OVX in a protein kinase A (PKA)-dependent way; this effect was reverted after E2 treatment [
66,
99]. Likewise, in cardiomyocytes from OVX guinea pigs, NCX activity was increased by 20%, and this effect was reverted with E2 treatment [
66,
100]. Moreover, E2 supplementation has been found to have a cardioprotective effect in ischemia/reperfusion injury models [
101].
In this sense, NCX overexpression models after myocardial infarction (MI) caused an overload in [Ca
2+]i in male mice but not in female mice. Male mice cardiomyocytes, when exposed to E2 (nanomolar range), decreased [Ca
2+]i post-MI in a concentration-dependent manner [
101]. Interestingly, in a group of female transgenic mice, the NCX overexpression did not lead to [Ca
2+]i overload, indicating the protective role of E2 to compensate for the greater activity of NCX [
101]. These findings point out that E2 exerts a protective function in cardiac myocytes. Indeed, further research clarified that post-MI E2-confered protection was mediated by NCX. In another study, it was confirmed that the myocardial contractile function (left ventricular developed pressure, dP/dt
max, dP/dt
min) in male transgenic NCX overexpression mice was significantly higher than in their WT counterparts, as well as in OVX transgenic females, but not in the transgenic or SHAM transgenic female groups. These results implicate NCX in both the contractile and relaxation aspects of the heartbeat. In the post-MI/reperfusion injury phase, the function recovery in transgenic males was lower than in WT males; however, female WT and transgenic NCX mice had a similar recovery, compared with the significantly diminished recovery in the OVX group. This post-ischemic functional recovery pattern coincides with the lower recovery of energy metabolites (ATP and phosphocreatine) as well as the alternans (heartbeats of alternating large and small amplitude at equal intervals) observed only in the hearts of the male transgenic and female OVX transgenic mice, corresponding to [Ca
2+]i overload. These findings suggest a protective role of estrogen over the NCX activity during ischemic/reperfusion injury [
102].
Furthermore, E2′s protective capacities were also observed in neurons, where nanomolar concentrations of E2 exerted rapid effects over the NCX function, increasing the outward Ca
2+ current and decreasing the Ca
2+ influx mediated by NCX; this effect was potentiated by insulin-like growth factor 1 (IGF-1) [
103]. These effects seem to be independent of the canonical estrogen signaling pathways, since the presence of an inhibitor of estrogen receptors (ICI182780, 10 µM) did not alter the results. The non-genomic activity of E2 over NCX in neurons leads to maintaining [Ca
2+]i at lower levels, preventing the activation of Ca
2+-dependent apoptosis. These mechanisms could be especially useful to counteract the cytotoxicity induced by glutamate through NMDA or AMPA receptor activation [
103,
104].
6. Plasma Membrane Ca2+ ATPase
To restore [Ca
2+]i to basal levels after increases induced by agonist stimulation, Ca
2+ can be pumped across the membrane and out of the cell against its electrochemical gradient, expending ATP during the process [
23,
24,
37,
105]. This mechanism is achieved by the ASM plasma membrane ATPase (PMCA) that has four different isoforms: PMCA1, -2, -3 and -4, but only PMCA1 and PMCA4 are expressed in this tissue (
Figure 3) [
24,
105]. PMCA activity in ASM cells has been implicated in many intracellular processes, including contraction regulation [
24], cellular proliferation [
105], and even apoptosis [
105]. It is possible that dysfunction of this protein could lead to an increase in [Ca
2+]i and favor AHR [
24,
105].
Modulation of PMCA in ASM cells by estrogen, either through genomic or non-genomic actions, has not been described (
Figure 3). However, this phenomenon has been studied in other cellular types. For instance, in prepubescent female rats treated for 3 days with E2 (40 µg/kg/day), the PMCA1 expression in the esophagus was decreased [
95]. In MCF-7 cells (breast cancer cell line), PMCA4b isoform expression was increased by E2 (1 nM) treatment in a way mediated by ERα [
106]. Conversely, E2 treatment for 24 h decreased the expression of the isoforms PMCA2 and -4 in human fibroblast-like synovial cells (HFLS) and in mouse macrophage-like cells in a dose-dependent manner [
107]. In another study, E2 exposure for 24 h did not alter PMCA expression in distal tubule kidney cells; however, PMCA activity was significantly enhanced [
108]. In human endometrium, PMCA1 expression was significantly upregulated when treated with E2 (physiological range) for 48 h [
56], compared to the decrease of PMCA1 expression in mouse uterus following E2 treatment [
57]. E2 has also been described to participate in mechanical pain sensitivity through PMCA2. In female mice, OVX increased mechanical pain sensitivity through PMCA2; this was reversed with E2 supplementation, in a ER-mediated way [
109]. It should be noted that alterations in the Ca
2+ machinery might participate in various pathophysiological processes in accordance with the type of cell implicated.
7. Sarcoplasmic Reticulum Ca2+ ATPase
Another essential Ca
2+-pump that participates in intracellular Ca
2+ reuptake that is located on the SR membrane of ASM cells is SERCA. It is in charge of driving Ca
2+ ions against its electrochemical gradient by ATP consumption to reestablish b[Ca
2+]i and restoring depleted Ca
2+ internal stores [
24,
37,
105]. Three isoforms of the SERCA protein are known: SERCA1, -2, and -3, with various alternative splicing isoforms. ASM expresses isoforms SERCA2a and 2b, which predominate [
24,
110,
111]. Alterations in SERCA expression or activity can lead to increases in [Ca
2+]i, a phenomenon that has been linked to an asthmatic ASM phenotype that contributes to airway remodeling and hyperresponsiveness [
24,
105,
111,
112,
113].
Interestingly, hASM cells exposed to E2 apparently showed no acute effects on SERCA activity (
Figure 3) [
20]. Contrastingly, the estrogen-induced genomic effects on SERCA were explored in hASM cells. They were treated with E2 (1 nM), ERα agonist (PPT, 10 nM) or an ERβ agonist (WAY, 10 nM) for 2 hrs, and then incubated with TNF-α or IL-13. The Ca
2+ response to histamine in the presence of TNF-α or IL-13 was significantly higher compared to the vehicle. A similar response was observed in the E2 and PPT groups, but the effect was reverted in the WAY group, that showed a response similar to the vehicle group. The time of [Ca
2+]i decay in the response induced by histamine was higher in the group treated with TNF-α or IL-13. This response was reversed by a treatment with WAY but not by E2 or PPT (
Figure 3) [
35]. These results were attributed to the treatment’s effects on SERCA2 expression, since there was a significant reduction in protein expression in the TNF-α or IL-13 treated cells, which was reverted in WAY treated cells but not in to those administered E2 or PPT (
Table 1) [
35].
The evidence indicates that [Ca2+]i can be modulated by estrogens by the coordinated participation of several target proteins, and that changes in Ca2+ availability induced by estrogens is dynamic and additive exerted through genomic and non-genomic events.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24097879