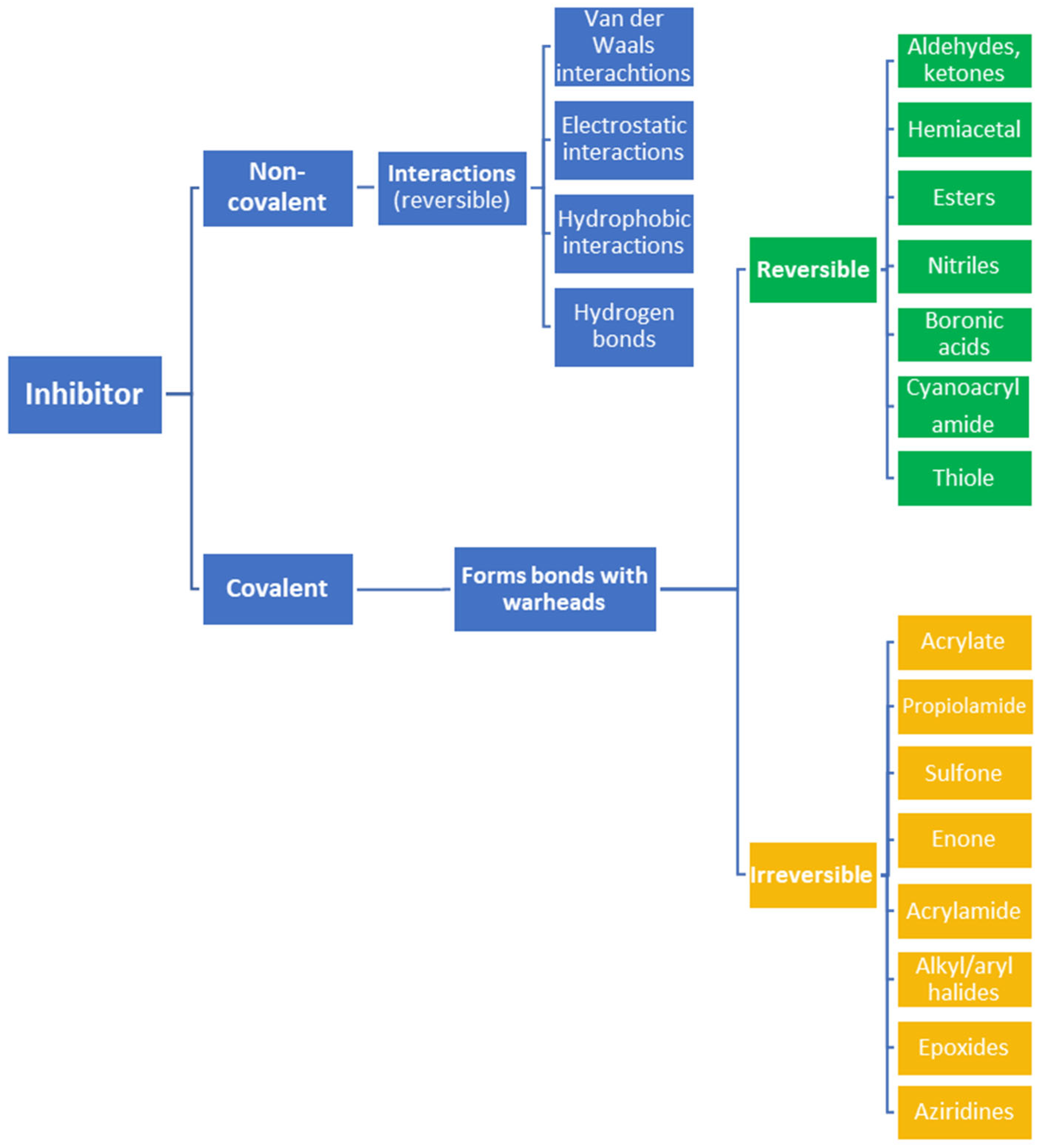
The formation of covalent bonds that target proteins can offer drugs diverse advantages in terms of target selectivity, drug resistance, and administration concentration. The most important factor for covalent inhibitors is the electrophile (warhead), which dictates selectivity, reactivity, and the type of protein binding (i.e., reversible or irreversible) and can be modified/optimized through rational designs. Furthermore, covalent inhibitors are becoming more and more common in proteolysis, targeting chimeras (PROTACs) for degrading proteins, including those that are currently considered to be ‘undruggable’.
- covalent inhibitors
- PROTACs
- drug discovery
- covalent drugs
1. Introduction
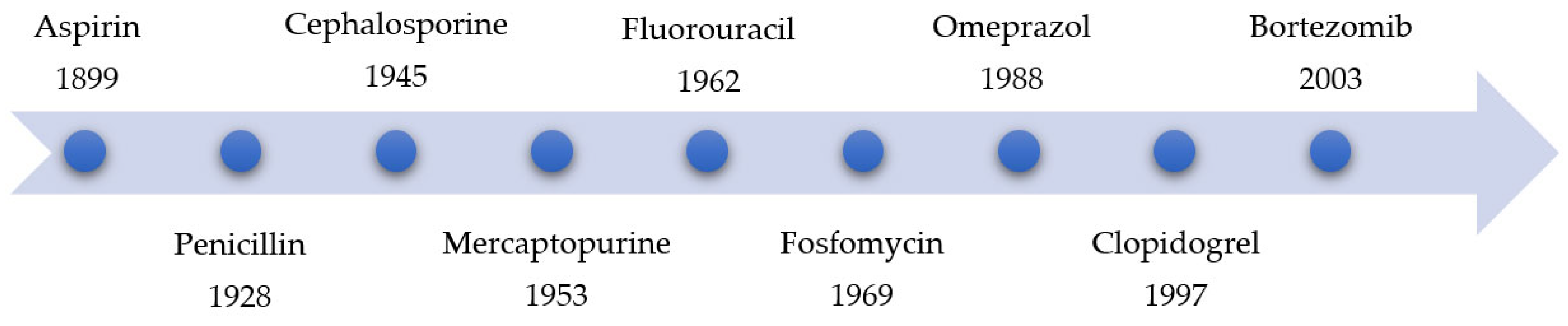
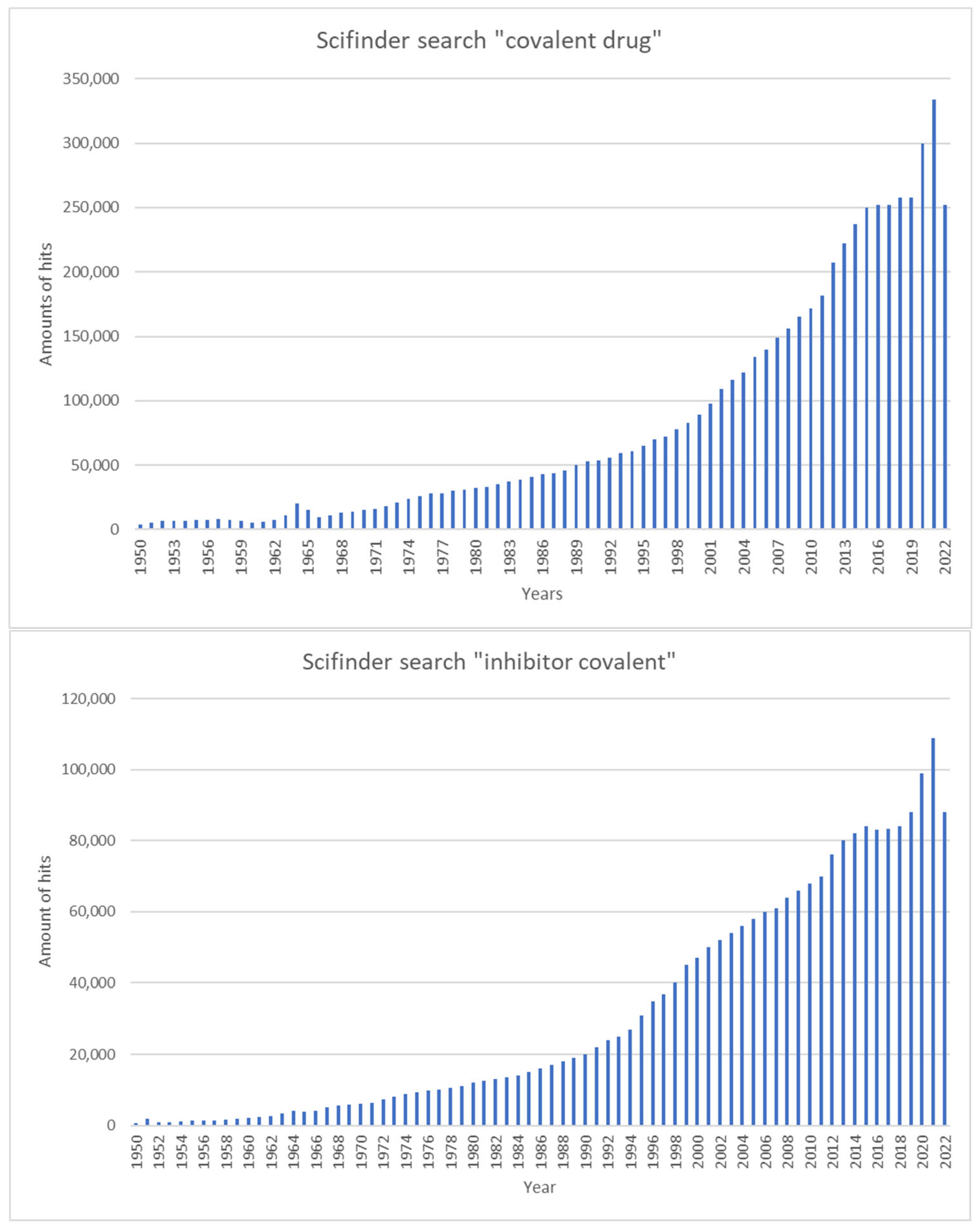
|
Year |
Name of Drug |
Warhead |
Function |
|---|---|---|---|
|
2010 |
Ceftaroline (β-lactam) |
|
β-lactam antibiotic |
|
2011 |
Telaprevir (α-ketoamide) |
|
HCV protease inhibitor |
|
2011 |
Boceprevir (α-ketoamide) |
|
HCV protease inhibitor |
|
2011 |
Abiraterone (-) |
- |
Prostate cancer treatment |
|
2012 |
Carfilzomib (epoxide) |
|
Proteasome inhibitor (cancer) |
|
2013 |
Afatinib (α,β-unsaturated carbonyl) |
|
EGFR tyrosine kinase inhibitor |
|
2013 |
Dimethyl fumarate (α,β-unsaturated carbonyl) |
|
Immunomodulatory drug |
|
2013 |
Neostigmine (carbonyl group) |
|
Acetylcholinesterase inhibitor |
|
2013 |
Ibrutinib (α,β-unsaturated carbonyl) |
|
EGFR tyrosine kinase inhibitor |
|
2014 |
Ceftolozane (β-lactam) |
|
β-lactam antibiotic |
|
2015 |
Osimertinib (α,β-unsaturated carbonyl) |
|
EGFR tyrosine kinase inhibitor |
|
2015 |
Olmutinib (α,β-unsaturated carbonyl) |
|
EGFR tyrosine kinase inhibitor |
|
2016 |
Narlaprevir (α-ketoamide) |
|
HCV protease inhibitor |
|
2017 |
Acalabrutinib (α,β-unsaturated propargylamide) |
|
Bruton’s tyrosine kinase inhibitor |
|
2017 |
Neratinib (α,β-unsaturated carbonyl) |
|
EGFR tyrosine kinase inhibitor |
|
2017 |
Vaborbactam (boronic acid) |
|
Non-β-lactam β-lactamase inhibitor |
|
2018 |
Dacomitinib (α,β-unsaturated carbonyl) |
|
EGFR tyrosine kinase inhibitor |
|
2019 |
Selinexor (α,β-unsaturated carbonyl) |
|
Nuclear export inhibitor |
|
2019 |
Zanubrutinib (α,β-unsaturated carbonyl) |
|
Bruton’s tyrosine kinase inhibitor |
|
2019 |
Cerfiderocol (β-lactam) |
|
β-lactam antibiotic |
|
2019 |
Voxelotor (aldehyde) |
|
Hemoglobin oxygen-affinity modulator |
|
2021 |
Sotorasib (α,β-unsaturated carbonyl) |
|
KRAS G12C inhibitor |
|
2021 |
Nirmatrevir (nitrile) |
|
SARS-CoV-2 main protease inhibitor |
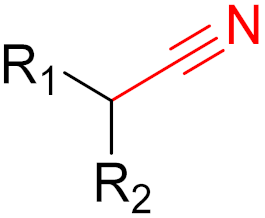
2. Advantages and Disadvantages of Covalent Inhibitors
|
Type of Inhibitor |
Advantages |
Disadvantages |
|---|---|---|
|
Non-Covalent |
|
|
|
Covalent |
|
|
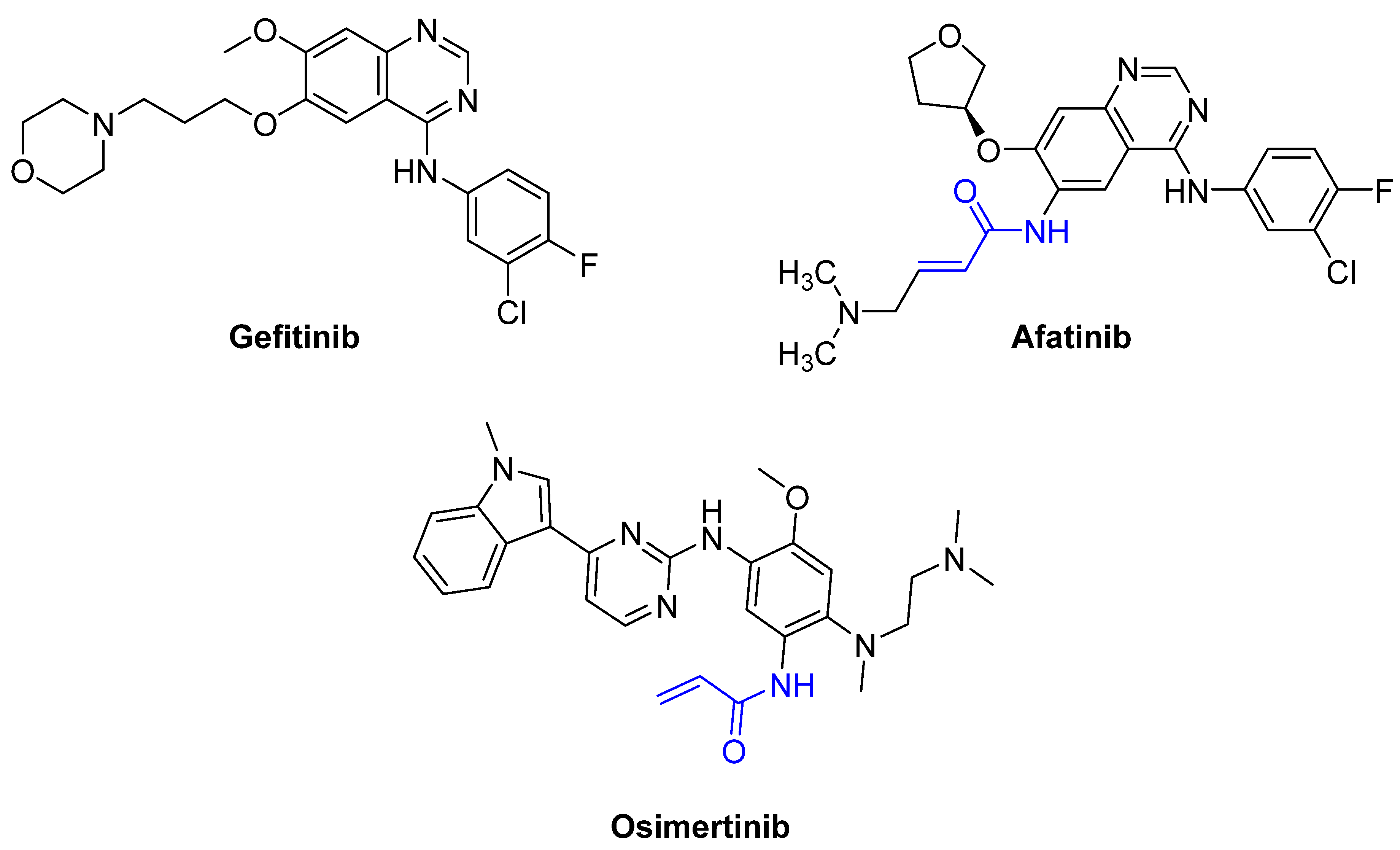
3. Mechanisms of Action
The entire process involving the interaction between a target and a covalent inhibitor up to the formation of a covalent bond takes place in two steps [6]. The first step is the reversible association between the inhibitor and the target protein [6]. In the second step, a reaction takes place that forms a covalent bond [6]. This is exemplified by telaprevir, which reversibly inhibits the viral NS3.4A protease of the hepatitis C virus (HCV; Figure 8) [32].
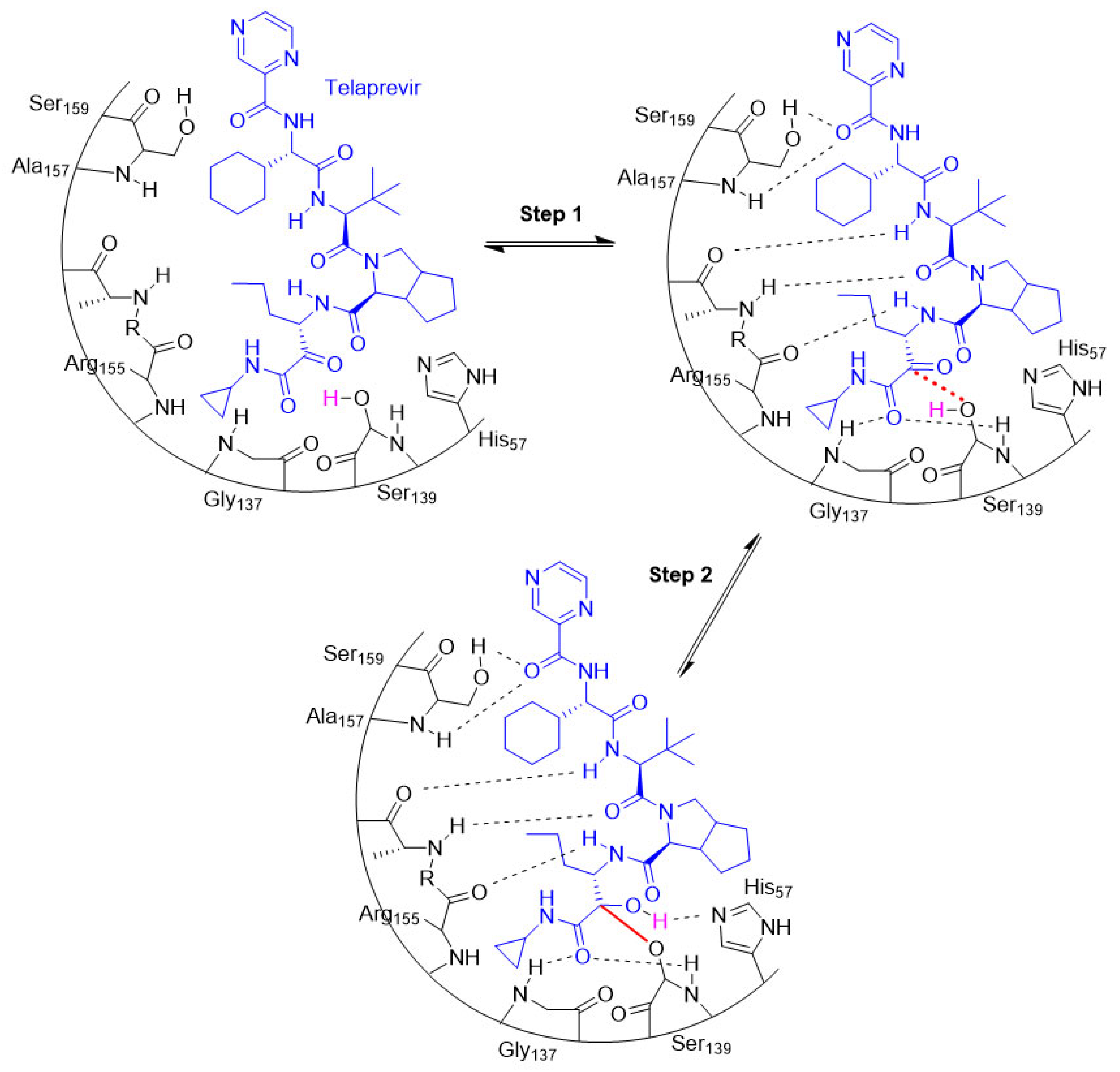
Figure 8. An illustration of the entire two-step process (i.e., association and bond formation) using telaprevir, the example HCV protease inhibitor. Telaprevir inhibits the viral NS3.4A protease of the hepatitis C virus.
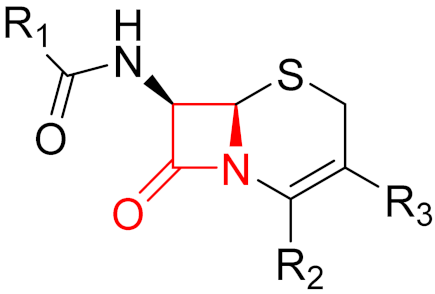
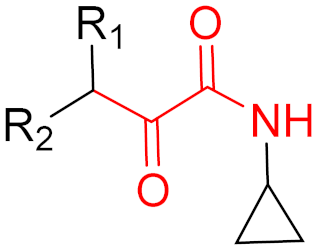
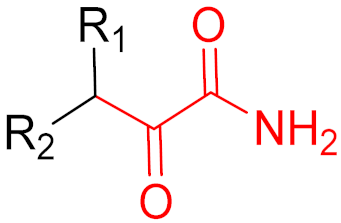
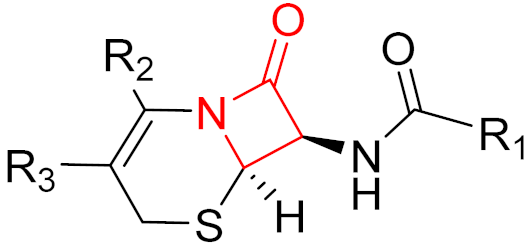
Often, covalent inhibitors carry electrophilic groups, which react with nucleophilic residue on the target enzymes [6][33]. The warheads can be epoxides, aziridines, esters, ketones, nitriles, or another similar group [33]. For example, penicillin is a covalent inhibitor with beta-lactam as the warhead, which reacts with the active serine residue in the D-alanine transpeptidase [11]. Transpeptidases are essential for cross-linking in the biosynthesis of bacterial cell walls [11]. Irreversible bonds inhibit transpeptidase, resulting in the lysis of bacterial cells due to their instability [11]. The mechanism of action of this irreversible inhibition is shown in Figure 9.
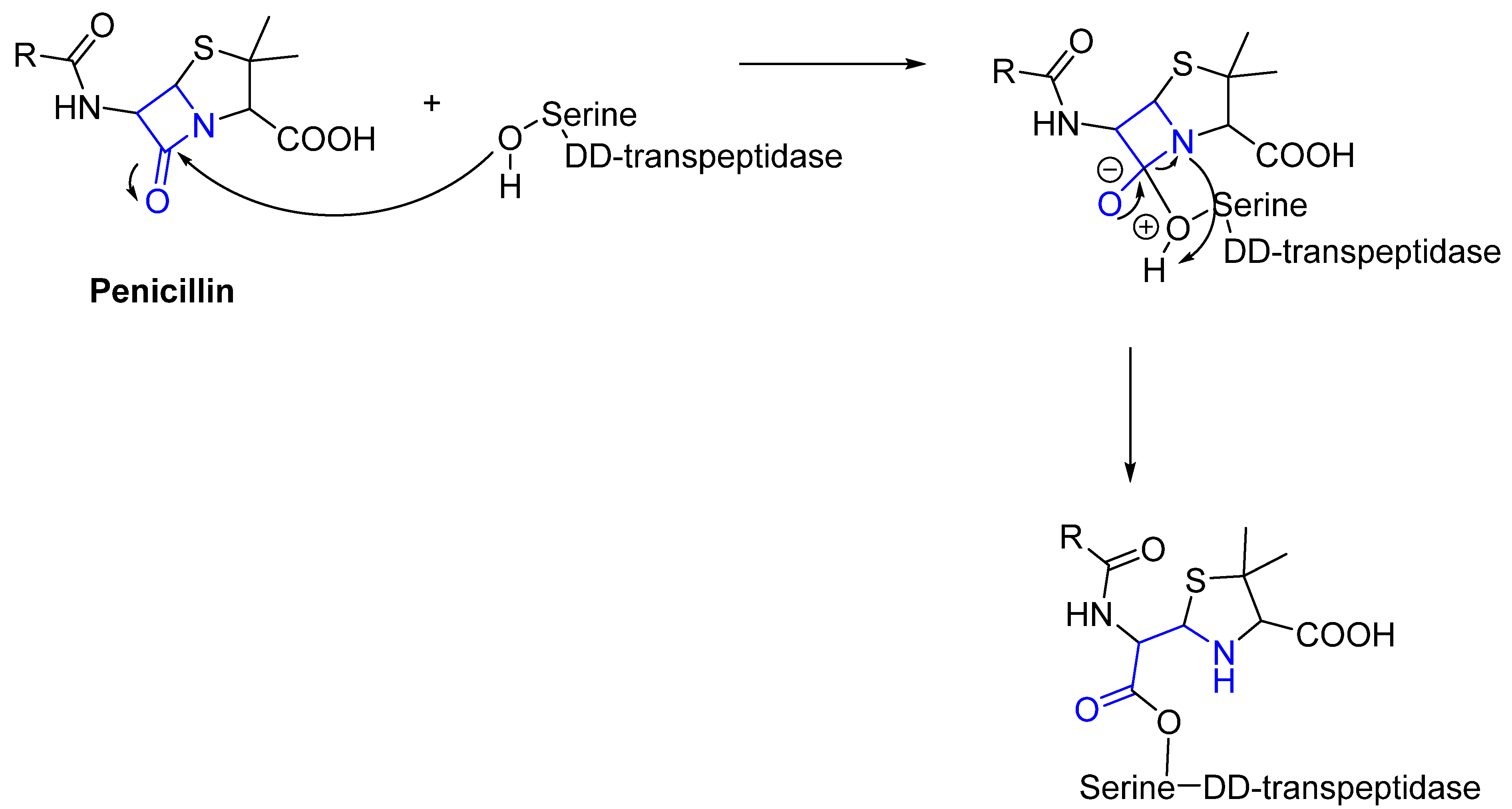
Figure 9. The mechanism of action of the irreversible inhibition of DD-transpeptidase by penicillin. The warhead of penicillin (β-lactam; highlighted in blue) reacts with the serine side chain of DD-transpeptidase.
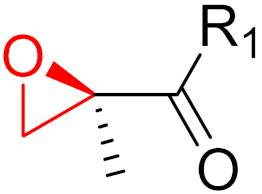
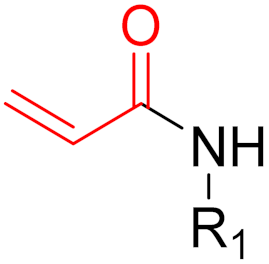
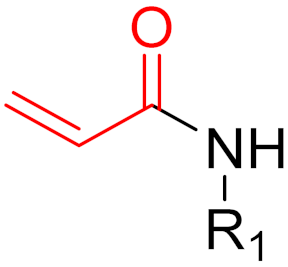
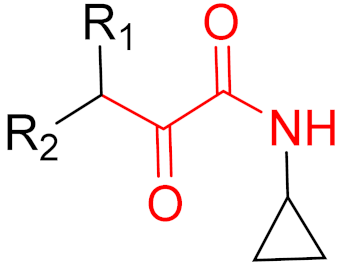
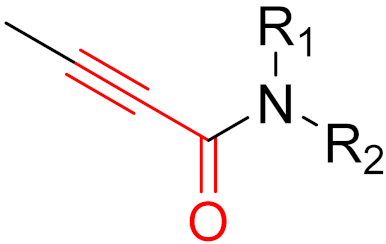
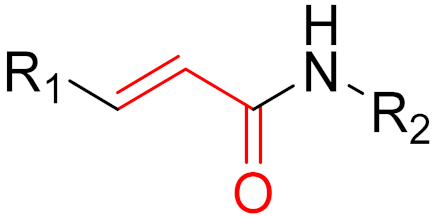
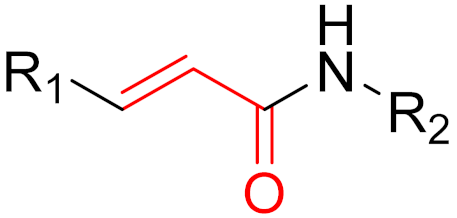
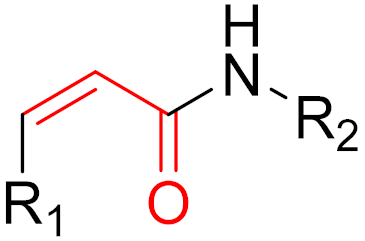
To date, many new functional groups have been found that can form covalent bonds with sulfur-containing functional groups, as shown in Figure 10 [34]. The advantage of these groups is that they can directly react with the cysteine in target proteins at its active site without a prior metabolic activation [34]. Figure 10 displays various warheads that are involved in the formation of irreversible and reversible bonds [34]. In most cases, inhibitors occupy the binding pockets, which prevents substrates from forming bonds (i.e., competitive inhibition) [34]. Nevertheless, occupation in the active sites of target proteins is not always necessary [34]. For instance, a few inhibitors can bind to the remote sides of target enzymes, resulting in the alternation of the binding pocket [34]. These inhibitors are called allosteric inhibitors [34]. In rare cases, uncompetitive inhibition can occur, in which inhibitors bind exclusively to enzyme–substrate complexes. This results in the formation of enzyme–substrate–inhibitor complexes, which ensures that the enzymes do not convert the substrates; therefore, no products are formed [35]. Irreversible inhibitors are divided into two types: affinity label inhibitors and mechanism-based inhibitors (suicide inhibitors) [36]. Affinity label inhibitors resemble enzyme substrates and enter the active sites of enzymes, where irreversible covalent bonds are formed, and the active sites are modified without enzymatic conversion [36]. Suicide inhibitors bind to active sites in the same way as substrates, triggering the enzymatic properties of the enzymes [36]. During the enzymatic process, intermediaries are formed that cannot be further converted or split off. As a result, no further substrates can be converted. Aspirin and penicillin are examples of suicide inhibitors [36].
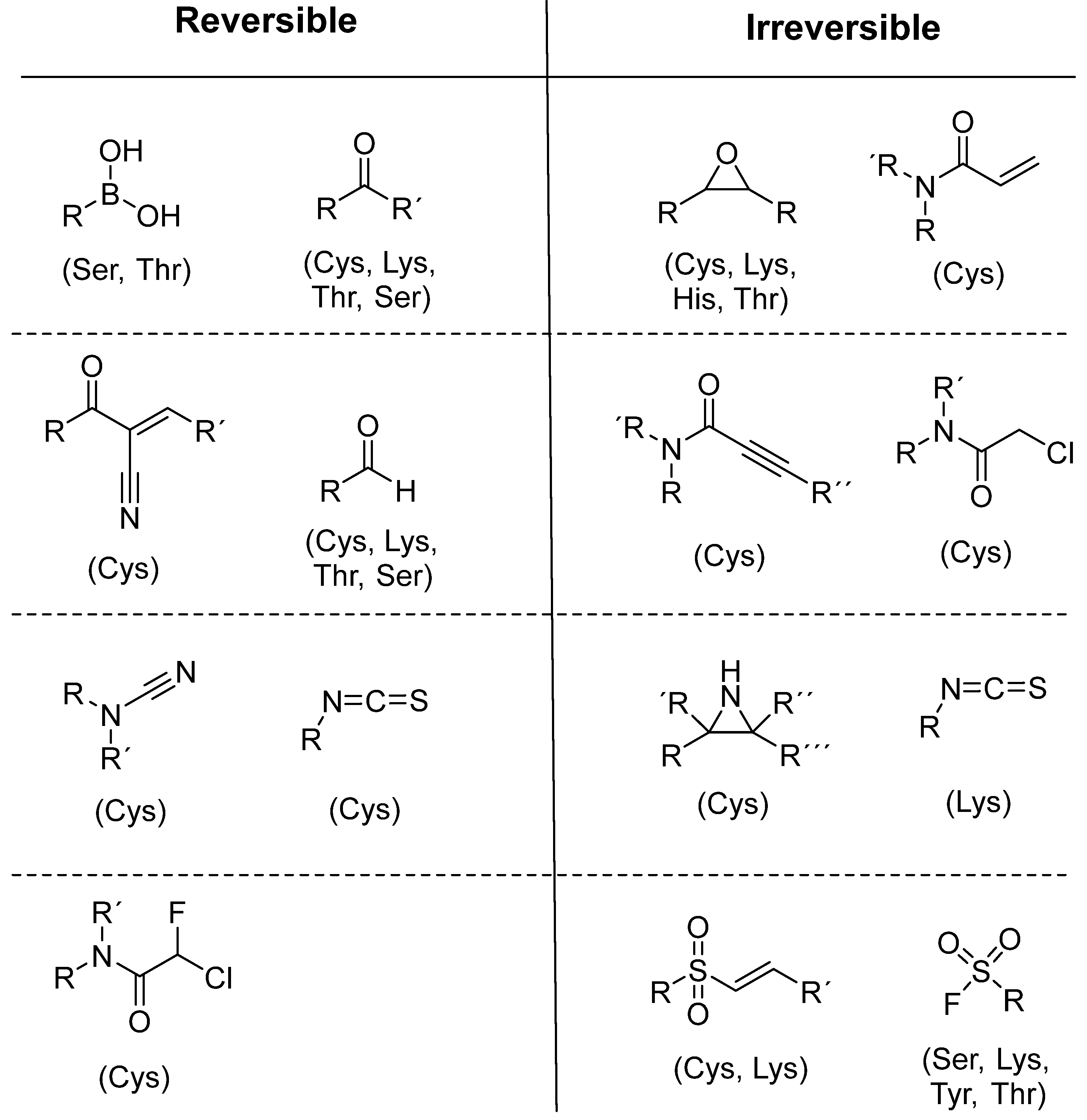
Figure 10. Warheads form irreversible and reversible bonds. The primary targeting amino acid residues are shown in brackets below the respective structures [37][38].
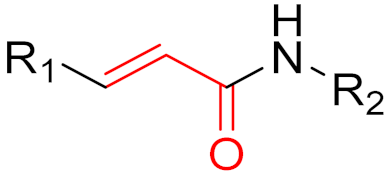
The formation of a bond can have different mechanisms. Many electrophilic warheads can react with a nucleophile via Michael addition, for example, [37]. The attacking nucleophile (e.g., carbanion, amine or thiol) serves as a Michael donor and the electrophile (e.g., α,β-unsaturated carbonyl compound) as a Michael acceptor [37]. A more detailed description of the different types of covalent reactions in drug development and the associated groups has been presented in great detail by Gehringer et al. [37] and Shindo et al. [38] and is not exclusively discussed here.
This entry is adapted from the peer-reviewed paper 10.3390/ph16050663
References
- Johnson, J.R.; Williams, G.; Pazdur, R. End points and United States Food and Drug Administration approval of oncology drugs. J. Clin. Oncol. 2003, 21, 1404–1411.
- Dumit, J. Drugs for Life: How Pharmaceutical Companies Define Our Health; Duke University Press: Durham, NC, USA, 2012.
- Rowland, M.; Tozer, T.N. Clinical Pharmacokinetics/Pharmacodynamics; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2005.
- Aljoundi, A.; Bjij, I.; El Rashedy, A.; Soliman, M.E. Covalent versus non-covalent enzyme inhibition: Which route should we take? A justification of the good and bad from molecular modelling perspective. Protein J. 2020, 39, 97–105.
- Jollow, D.; Mitchell, J.; Potter, W.; Davis, D.; Gillette, J.; Brodie, B. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J. Pharmacol. Exp. Ther. 1973, 187, 195–202.
- Baillie, T.A. Targeted covalent inhibitors for drug design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421.
- Jack, D.B. One hundred years of aspirin. Lancet 1997, 350, 437–439.
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317.
- Baillie, T.A. The contributions of Sidney D. Nelson to drug metabolism research. Drug Metab. Rev. 2015, 47, 4–11.
- Lei, J.; Zhou, Y.; Xie, D.; Zhang, Y. Mechanistic Insights into a Classic Wonder Drug Aspirin. J. Am. Chem. Soc. 2015, 137, 70–73.
- Fleming, A. Penicillin. Br. Med. J. 1941, 2, 386.
- Kumalo, H.M.; Bhakat, S.; Soliman, M.E. Theory and applications of covalent docking in drug discovery: Merits and pitfalls. Molecules 2015, 20, 1984–2000.
- Vane, J.; Botting, R. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258.
- Wang, Y.-H.; Zhang, F.; Diao, H.; Wu, R. Covalent Inhibition Mechanism of Antidiabetic Drugs—Vildagliptin vs Saxagliptin. ACS Catal. 2019, 9, 2292–2302.
- Smith, A.J.; Zhang, X.; Leach, A.G.; Houk, K. Beyond picomolar affinities: Quantitative aspects of noncovalent and covalent binding of drugs to proteins. J. Med. Chem. 2009, 52, 225–233.
- Adeniyi, A.A.; Muthusamy, R.; Soliman, M.E. New drug design with covalent modifiers. Expert Opin. Drug Discov. 2016, 11, 79–90.
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent inhibition in drug discovery. ChemMedChem 2019, 14, 889–906.
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792.
- Nakamura, Y.; Oka, M.; Soda, H.; Shiozawa, K.; Yoshikawa, M.; Itoh, A.; Ikegami, Y.; Tsurutani, J.; Nakatomi, K.; Kitazaki, T. Gefitinib (“Iressa”, ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, reverses breast cancer resistance protein/ABCG2–mediated drug resistance. Cancer Res. 2005, 65, 1541–1546.
- Koehler, J.; Schuler, M. Treatment, Afatinib, erlotinib and gefitinib in the first-line therapy of EGFR mutation-positive lung adenocarcinoma: A review. Oncol. Res. 2013, 36, 510–518.
- Dungo, R.T.; Keating, G.M. Afatinib: First global approval. Drugs 2013, 73, 1503–1515.
- Singh, J. The Ascension of Targeted Covalent Inhibitors. J. Med. Chem. 2022, 65, 5886–5901.
- Greig, S.L. Osimertinib: First Global Approval. Drugs 2016, 76, 263–273.
- Shaikh, M.; Shinde, Y.; Pawara, R.; Noolvi, M.; Surana, S.; Ahmad, I.; Patel, H. Emerging Approaches to Overcome Acquired Drug Resistance Obstacles to Osimertinib in Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65, 1008–1046.
- Cohen, S.D.; Pumford, N.R.; Khairallah, E.A.; Boekelheide, K.; Pohl, L.R.; Amouzadeh, H.; Hinson, J.A. Selective protein covalent binding and target organ toxicity. Toxicol. Appl. Pharmacol. 1997, 143, 1–12.
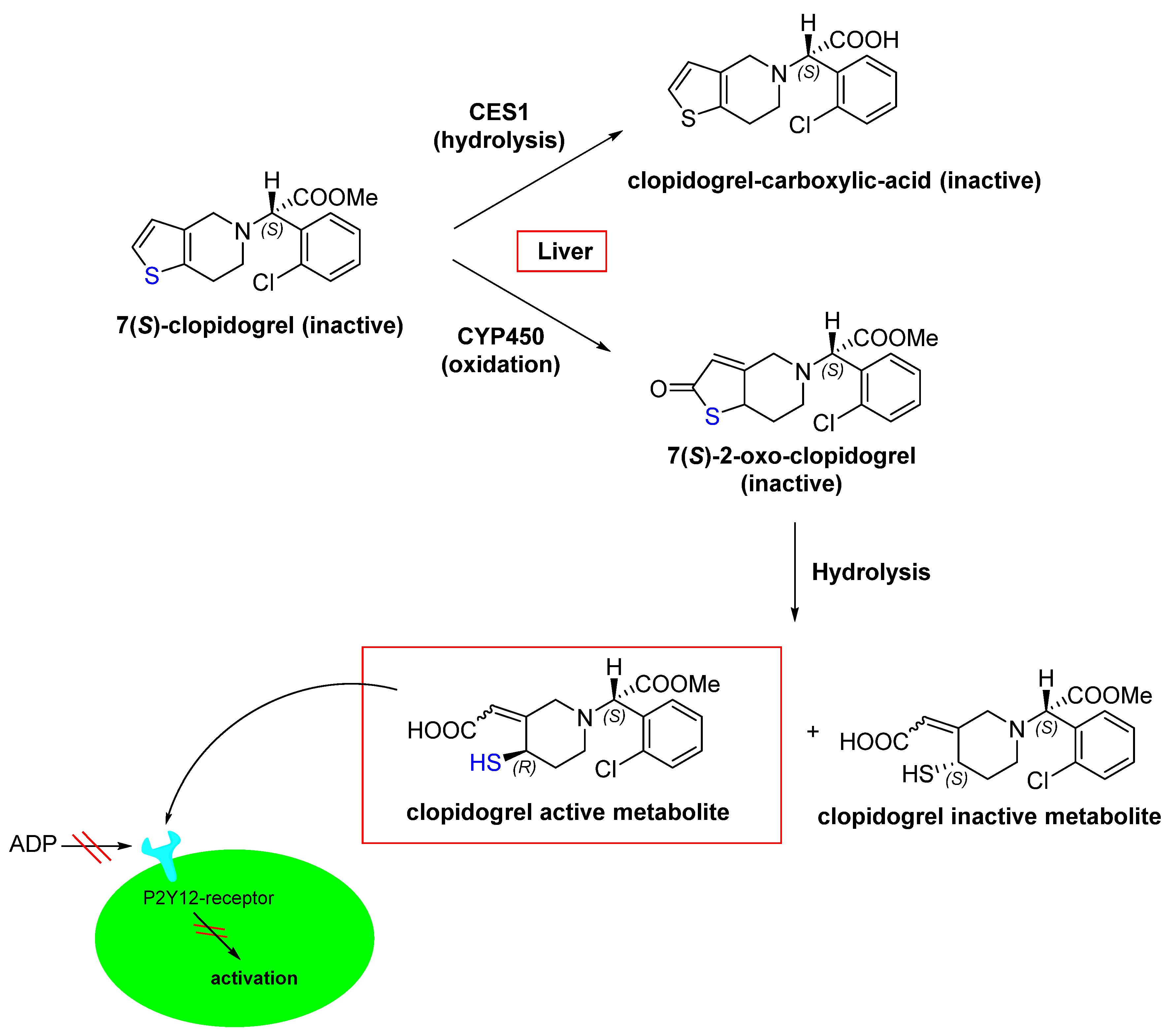
- Coukell, A.J.; Markham, A. Clopidogrel. Drugs 1997, 54, 745–750.
- Jarvis, B.; Simpson, K. Clopidogrel. Drugs 2000, 60, 347–377.
- Bryant, J.; Post, J.M.; Alexander, S.; Wang, Y.-X.; Kent, L.; Schirm, S.; Tseng, J.-L.; Subramanyam, B.; Buckman, B.; Islam, I. Novel P2Y12 adenosine diphosphate receptor antagonists for inhibition of platelet aggregation (I): In vitro effects on platelets. Thromb. Res. 2008, 122, 523–532.
- Bluet, G.; Blankenstein, J.; Brohan, E.; Prévost, C.; Chevé, M.; Schofield, J.; Roy, S. Synthesis of the stabilized active metabolite of clopidogrel. Tetrahedron 2014, 70, 3893–3900.
- Shringarpure, R.; Davies, K.J.A. Protein turnover by the proteasome in aging and disease. Free Radic. Biol. Med. 2002, 32, 1084–1089.
- Davies, A.; Nixon, A.; Muhammed, R.; Tsintzas, K.; Kirkham, S.; Stephens, F.B.; Moran, G.W. Reduced skeletal muscle protein balance in paediatric Crohn’s disease. Clin. Nutr. 2020, 39, 1250–1257.
- Perry, C.M. Telaprevir. Drugs 2012, 72, 619–641.
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073.
- Johnson, L.N.; Noble, M.E.; Owen, D.J. Active and inactive protein kinases: Structural basis for regulation. Cell 1996, 85, 149–158.
- Ouertani, A.; Neifar, M.; Ouertani, R.; Masmoudi, A.S.; Mosbah, A.; Cherif, A. Effectiveness of enzyme inhibitors in biomedicine and pharmacotherapy. Adv. Tissue Eng. Regen. Med. Open Access 2019, 5, 85–90.
- Hajizadeh, M.; Moosavi-Movahedi, Z.; Sheibani, N.; Moosavi-Movahedi, A.A. An outlook on suicide enzyme inhibition and drug design. J. Iran. Chem. Soc. 2022, 19, 1575–1592.
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724.
- Shindo, N.; Ojida, A. Recent progress in covalent warheads for in vivo targeting of endogenous proteins. Bioorg. Med. Chem. 2021, 47, 116386.