Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Multicomponent reactions are a fascinating family of organic chemistry transformations. Traditional bimolecular reactions are outperformed by such reactions, which combine three or more reactants into one reaction product. Multicomponent reactions speed up chemical space exploration by minimizing the quantity of synthesis and refinement steps needed to create a particular target. Isocyanides (isonitriles) were the only stable organic molecules containing a formally divalent carbon atom for a long period of time. The group of isocyanides are distinguishable from other functional groups due to their reactivity.
- isocyanide
- multicomponent reactions
- antibiotic resistance
1. Introduction
Multicomponent-based reactions are mainly chemical reactions where three or more compounds are used to make a final product. It has been almost 150 years since the world of chemistry was introduced to multicomponent reactions. Multicomponent reactions are a fascinating family of organic chemistry transformations [1]. Traditional bimolecular reactions are outperformed by such reactions, which combine three or more reactants into one reaction product [1]. Multicomponent reactions speed up chemical space exploration by minimizing the quantity of synthesis and refinement steps needed to create a particular target [2]. The associated atom economy of multicomponent reactions improves the chemical enterprise’s long-term viability even further.
Multicomponent reactions (MCRs) provide valuable methods for creating small-molecule compound libraries and are essential for studying structure–activity relationships (SARs). Since a number of multicomponent reactions produce exceptional scaffolds, the capacity to further qualify or functionalize them is critical for determining the scaffold’s biological value. Many of these scaffolds have a distinctive structure that allows them to investigate biological targets that regular scaffolds cannot. Novel scaffolds are becoming increasingly sought after for the treatment of infections that are liable to become unsusceptible to conventional treatments, and inventing anti-aging compounds that are required to combat diseases and conditions such as Parkinson’s disease, diabetes, Alzheimer’s, and cancer.
2. Isocyanides and the Types of Isocyanide-Based Multicomponent Reactions
Isocyanides (isonitriles) were the only stable organic molecules containing a formally divalent carbon atom for a long period of time. The group of isocyanides are distinguishable from other functional groups due to their reactivity. All commercially available isocyanides are volatile and emit a foul, harsh, and repulsive odor. It has been examined as a potential non-lethal weapon as a result of this type of odor. There are many isocyanide-based multicomponent reactions, the majority of which reportedly assist in chemical synthesis through using UGI’s four-component-based reaction. Though, in the following contexts, the distinct types of isocyanide-based multicomponent reactions are described.
In 1859, Lieke discovered isocyanides from the random reaction of silver cyanide and allyl iodide [3]. Later, Gautier and Hoffmann coined the word “isonitrile” after synthesizing these compounds by experimenting primary amines with alkali and chloroform [4]. Passerini presented the first IMCRs around half a century later, in 1921. Passerini’s three-component reaction (P-3CR) between an isocyanide, a carbonyl molecule, and a carboxylic acid resulted in the formation of α-acyloxy carboxamide 4. Although, attempts have been made to produce isocyanides in a more practical manner since the 1950s [5].
Finally, U-4CR began in the middle of 19th century, when Ivar Karl Ugi (1930–2005) synthesized α-acylamido carboxamide 9 by a single reaction of amine, carbonyl molecule, isocyanide, and acid [6][7][8]. The reaction has been referred to as the Ugi response since 1962. Around 20 years later, the modification of this reaction was carried out on a solid phase [9]. A research team has achieved the first stereoselective Ugi reaction on the solid phase in 2000 [10]. Finally, GBB-3CR, product 13 was discovered as the most recent MCR [11][12][13].
2.1. The Reactions of Passerini
Mario Passerini created the Passerini reaction in 1921. It was the first MCR to incorporate isocyanide and continues to play a significant role in combinatorial chemistry today. It entails the use of an aldehyde or ketone, an isocyanide, and a carboxylic acid, and provides direct access to -hydroxy carboxamides (Figure 1: Reaction 1). Ugi’s hypothesis indicates a non-ionic pathway, as the reaction is expedited in aprotic solvents (Figure 1: Reaction 2) [14]. The isocyanide’s nucleophilic attack follows the electrophilic activation of the carbonyl group. This produces a nitrilium intermediate, which is later targeted by carboxylate.
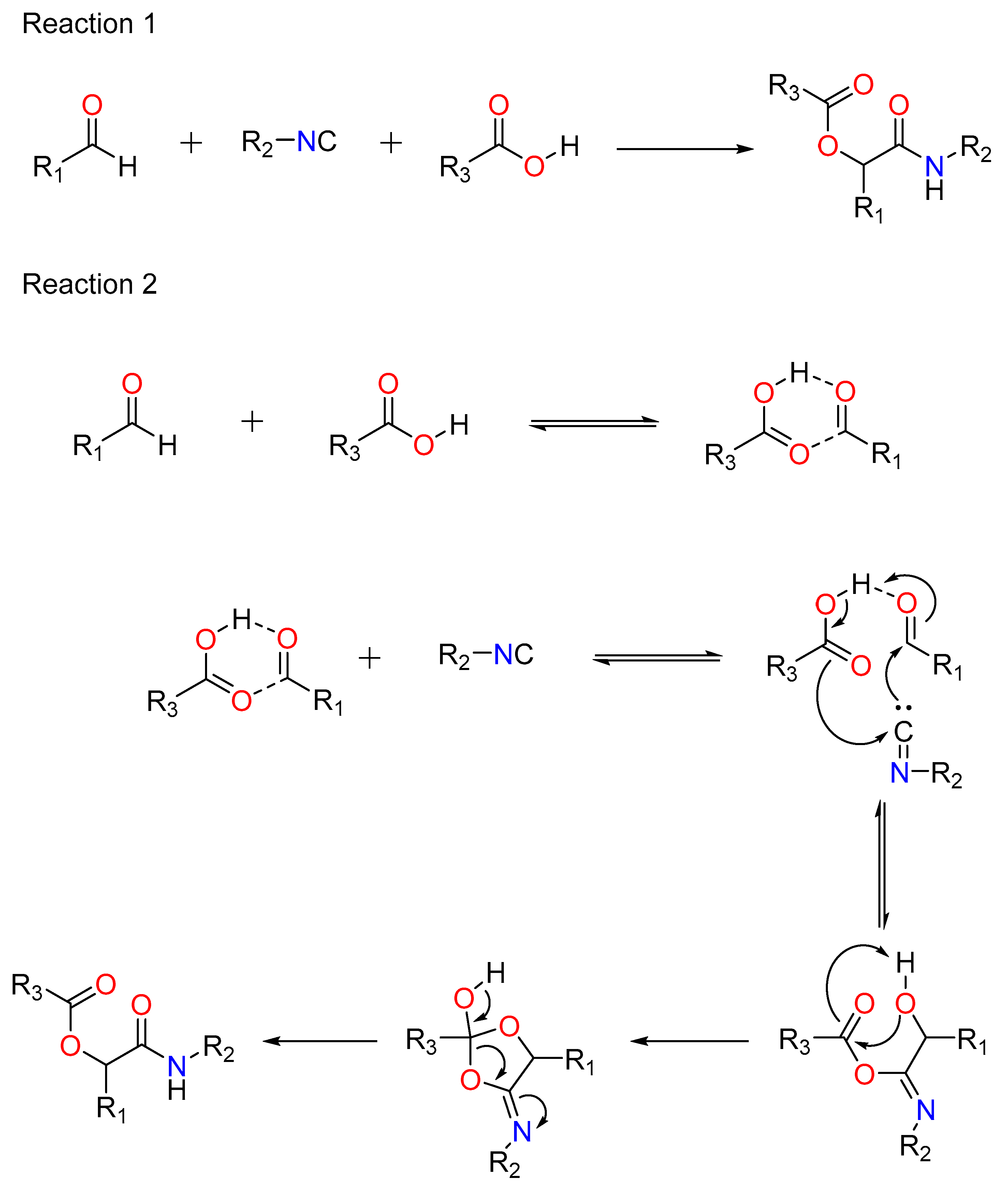
Figure 1. Reaction 1: A general Passerini reaction yielding an α-acyloxy amide, Reaction 2: The mechanism of Passerini’s general reaction.
Following on from Passerini’s general reaction, many scientists have experimented with different compounds. Recently, research by Wang et al. has included asymmetric reactions using widely available chiral Lewis acids [15]. The Zhu lab has documented Passerini reactions with alcohols, isocyanides, and carboxylic acids, broadening the reaction’s potential utility beyond carbonyl-containing molecules. The process employs catalytic TEMPO, CuCl2, and NaNO2 to convert an alcohol to an aldehyde [16].
Passerini-type reactions between free alcohols (isopropanol), aldehydes (unsaturated and aryl), and isocyanides (such as t-butyl isocyanide) have been reported in the presence of In (III) [17]. El Kam and Grimaud’s pioneering study resulted in the revelation of what is now known as the Passerini–Smiles reaction.
In this situation, the carboxylic acid is replaced by an electron-deficient phenol, such as 2-nitrophenol (or other nitrogen heteroaromatic but electron-deficient phenols). This method is thought to include stimulation of the aldehyde by the frail acidic phenol (pKa ~4.2), rendering the carbonyl electrophilic and susceptible to attack by the isocyanide. The phenol attacks the incipient nitrilium ion, followed by a SNAr, forming an -aryloxy amide. The critical step, according to current thinking, is the irreversible Smiles rearrangement of the intermediate phenoxyimidate adduct (Figure 2) [18].
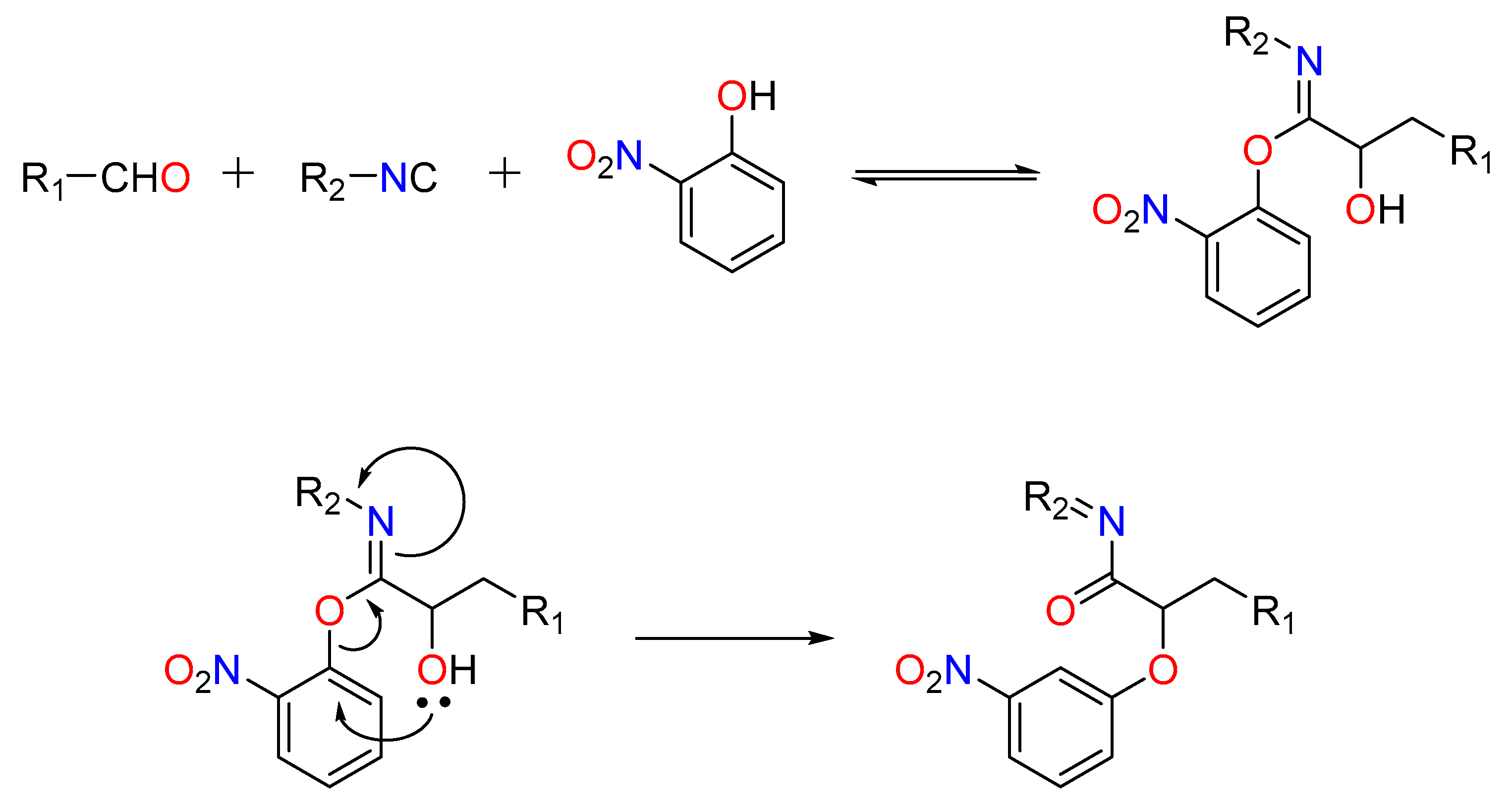
Figure 2. The Passerini–Smiles reaction.
A relatively recent but significant addition made by Soeta et al. is the substitution of silanols for carboxylic acids in the Passerini reaction, allowing for the production of α--siloxyamides. The mechanism involves the coordination of the silyl group to the carbonyl’s oxygen. This makes it vulnerable to nucleophilic attack by an isocyanide, which is followed by the intramolecular trapping of the nitrilium ion by the silanol’s alcoholic functional group [19] (Figure 3).
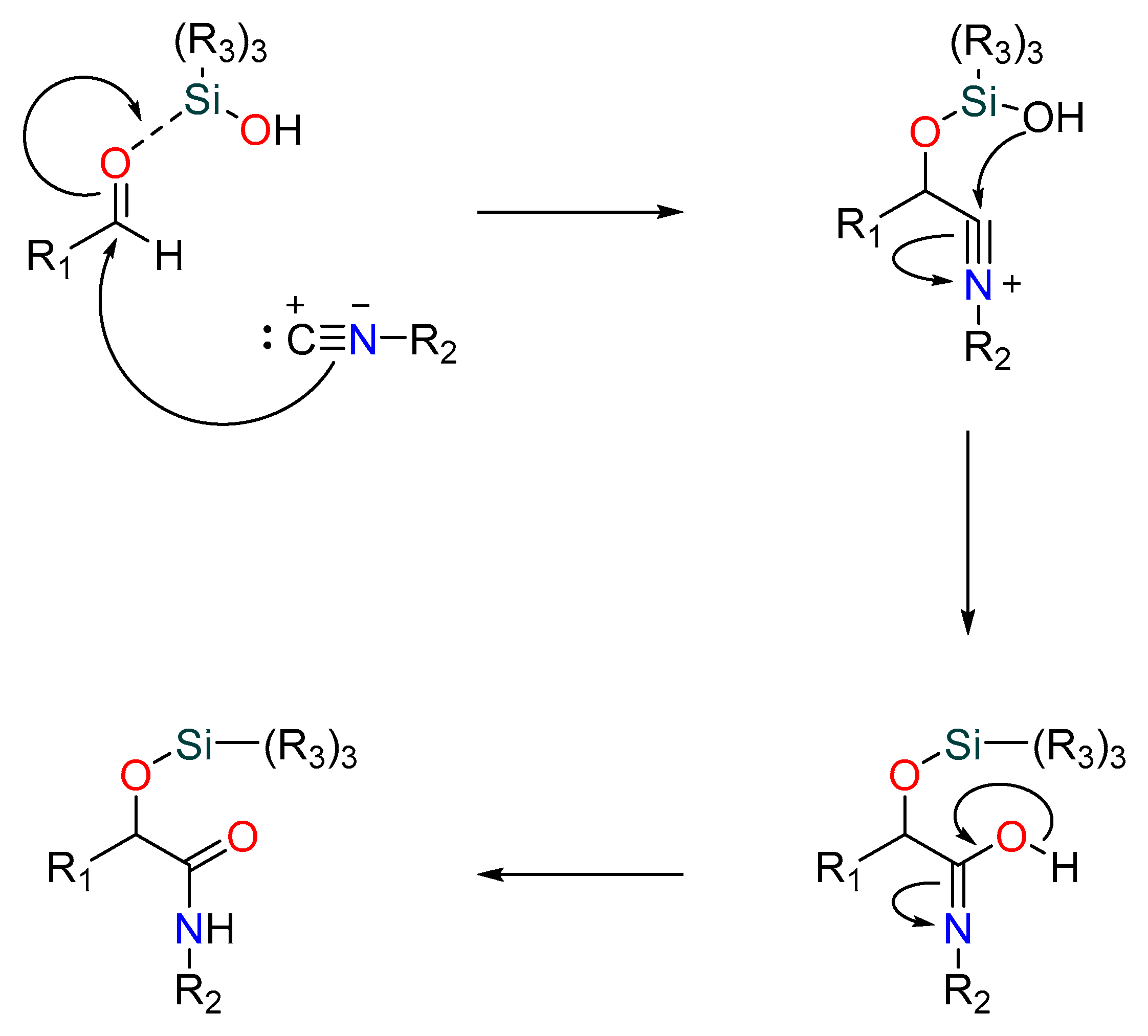
Figure 3. Passerini reaction yielding α-(sulfonyloxy) amides.
2.2. Ugi-4C Reaction
A ketone or aldehyde, a carboxylic acid, an isocyanide, and an amine are used in a traditional Ugi four-component reaction (U4CR).Typically, the reaction is carried out in high concentrations of methanol or 2,2,2-trifluoroethanol. The first step involves creating an imine by reacting the amine with the carbonyl compound, followed by the isocyanide’s nucleophilic attack, which produces the highly reactive nitrilium intermediate. The carboxylic acid then attacks the nitrilium, resulting in the formation of a central bis-amide via intramolecular Mumm rearrangement (Figure 4).
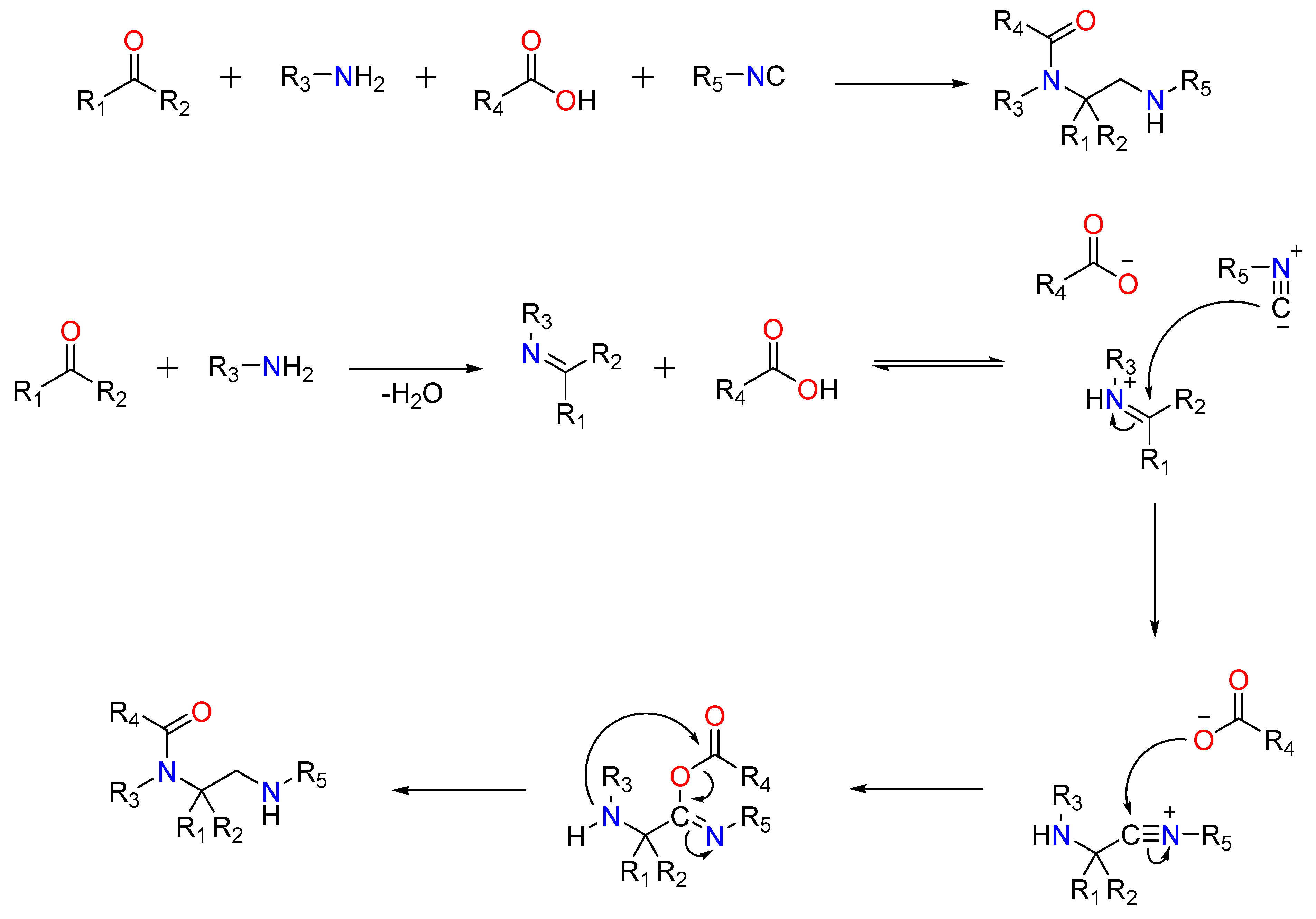
Figure 4. The Traditional Ugi-4C Component Reaction and The mode of action of the Ugi-4C Reaction.
Post-Ugi reactions have been reported depending on the R groups. Among the most notable are the Ugi-Heck, Ugi-Diels-Alder, Ugi-click, and Ugi-Buchwald-Hartwig reactions, in which a Ugi bis-amide containing reactive functional groups undergoes secondary reactions to form a ring. On the other hand, linear bis-amides are useful for the synthesis of peptides (both linear and cyclic) and peptidomimetics (Figure 4) [20][21][22][23][24].
2.3. Groebke–Blackburn–Bienaymé Reaction
In a non-concerted [4 + 1] reaction between an imine generated through the reaction between an aldehyde and an amine and an isocyanide, the reaction gives 3-aminoimidizoles [13][25][26]. This is called the Groebke–Blackburn–Bienaymé reaction, as shown in Figure 5. The nitrilium ion is trapped by the heterocyclic nitrogen, resulting in the production of an imidazole ring after rearrangement [12][27]. Kinase inhibitors, topoisomerase II inhibitors, antibacterials efficient against methicillin-resistant Staphylococcus aureus, fluorescence probes, and HIV-1 reverse transcriptase inhibitors are among the bioactive molecules created using this method [28][29][30][31][32][33].
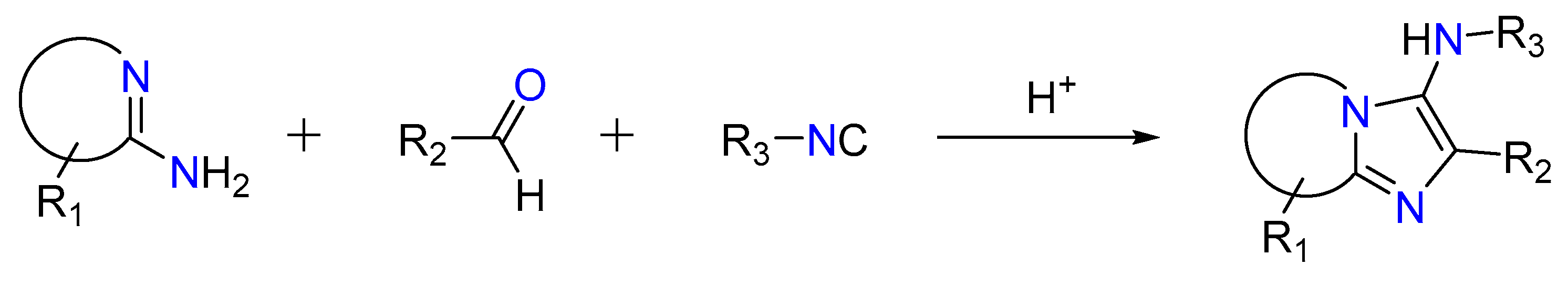
Figure 5. Groebke–Blackburn–Bienaymé reaction.
3. Groebke–Blackburn–Bienaymé Reaction in the Discovery of the Modified Antibiotic Trimethoprim
Antibiotic resistance is a new concern in public health. It does not only have high morbidity but also also has high mortality rates. Trimethoprim is a known antibiotic in the medicine field which is generally used with Sulfamethoxazole to treat urinary tract infections, which are caused by Staphylococcus aureus in cystic fibrosis patients, acute or severe bacterial diarrhea, or dysentery, and to protect infected areas from the opportunistic bacteria Pneumocystis carinii, which causes pneumonia in AIDS patients [34]. Combined medications of Trimethoprim and Sulfamethoxazole used to work by blocking two enzymes involved in the biosynthesis of folic acid: dihydropteroate synthetase and dihydrofolate reductase (DHFR), respectively [35]. Folate is produced by bacteria and is required for the biosynthesis of thymidine, which is required for DNA synthesis. As a result, when these antibiotics are taken together, they have a synergistic impact, limiting the growth of bacteria and eventually forwarding towards cell death. Later, when bacteria developed resistance to this treatment, determining progressive drugs to combat multidrug-resistant infections and organisms became increasingly difficult, as is the case with multidrug-resistant Pseudomonas aeruginosa and methicillin-resistant Staphylococcus aureus (MRSA). Combined medication of the two antibiotics has been found to be ineffective in treating infections caused by bacteria that have TMP-resistant DHFR enzymes. Many research teams have presented chemical variability at the residue of trimethoxybenzyl of the trimethoprim for the optimization of the medicine’s characteristics, overall activity, and to address trimethoprim resistance issues, resulting in the discovery of potential compounds against E. coli and S. aureus [36][37][38][39]. In 2019, Pedrola and her research team published a study wherein they modified the 2,4-diaminopyrimidine moiety of the drug Trimethoprim by the application of Groebke–Blackburn–Bienaymé reaction. In their study, they used the GBBR to create a series of TMP derivatives by fostering interactions between the trimethoprim and distinct types of ketones, aldehydes, and isocyanides, and then analyzed the resultant MCR compound as new antibacterial agents, determining their efficiency and potency while also taking into account their possible effect on resistant bacteria [40].
TMP has undergone chemical alterations based on the newly discovered effect of GBBRs on diaminopyrimidines, which entail selective and numerous MCRs [41]. The trimethoprim analogs therefore comprise a regioselective mono Groebke–Blackburn–Bienaymé reaction with an isocyanide/aldehyde pair, yielding by-products. It is worth noting that the advantageous production of the discovered isomer is justified by a kinetic control. Double GBBR procedures on TMP also result in two secondary by-products, and both of them are equivalents to each reactant class. To adequately produce and initiate the imine intermediate and obtain a decent yield, a range of Lewis acid catalysts was used. In addition, to obtain the pure product, conventional flash chromatography purification was usually required. The TMP reactant’s imidazo-azine scaffolds (N-fused bicyclic) were employed in the analogs, showing the variance points at R1, which came from the isocyanide input. R2 came from the aldehyde reactant (Figure 6).
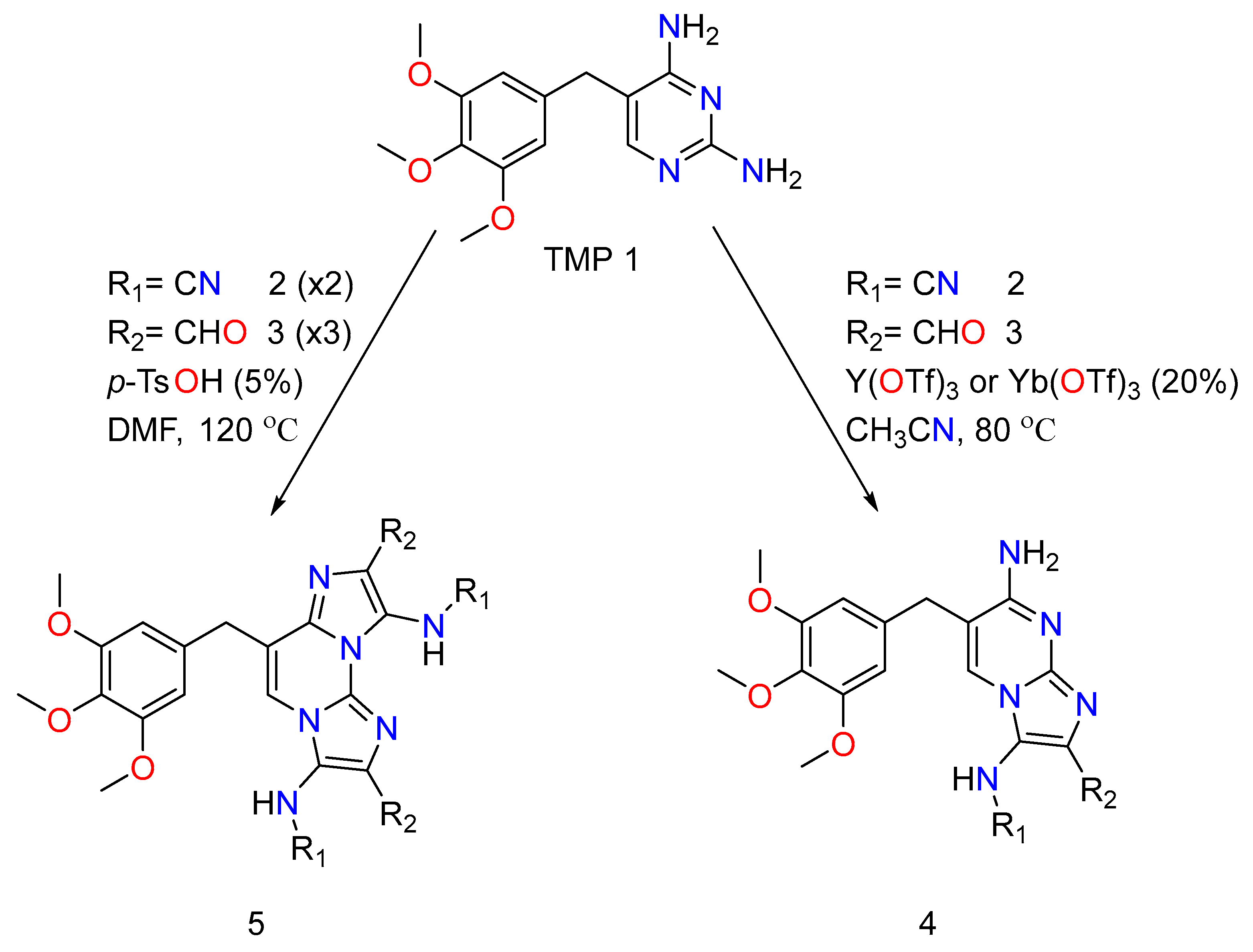
Figure 6. The chemical action of mono and double Trimethoprim GBBR compounds.
The procedures performed as expected in their TMP (Trimethoprim) system generate the desired compounds and exhibit similar reactivity and selectivity trends as the unsubstituted diaminopyrimidine experiments [41]. For primary screening, they have created an order of trimethoprim analogs that have a distinct effect on the imidazole amino group (R1 is 4-methoxyphenyl,ethoxycarbonylmethyl and cyclohexyl,tert-butyl), and a distinct variety of aromatic or alkyl substituents at the carbon position (R2 being methyl, α-, β-, or γ-pyridinyl, 4-chlorophenyl, isopropyl and α-thienyl). These reactions were prosperous, providing acceptable yields of mono-Groebke–Blackburn–Bienaymé by-products, and doubly substituted-Groebke–Blackburn–Bienaymé adducts. As a result, twelve novel compounds were derived, and the appropriate aldehyde/isocyanide combined mixture were synthesized as pure materials in this manner. Later, they decided to add an unsubstituted amino group to the imidazole ring of the new derivatives to help them be recognized by the DHFR function region, as the native substrate does. They then went about making such compounds by acidically removing a tert-butyl group from a suitable precursor adduct derived from MCRs involving tert-butyl isocyanide.
Though all of the abducts had MIC values greater than TMP (Trimethoprim) against S. aureus ATCC 29213 and E. coli ATCC 25922, some of them were almost as efficacious as TMP (Figure 7: 6–10). TMP, as well as all novel compounds, proved to be completely ineffective against P. aeruginosa PAO1 [42]. Almost all of the novel compounds, such as the control drug TMP, responded synergistically with SMX against E. coli ATCC 25922 and S. aureus ATCC 29213, with the other species being substantially more sensitive to the SMX combination than to the TMP–GBBR (Trimethoprim and Groebke–Blackburn–Bienaymé adducts) analogs alone. It was also discovered that nearly all of the novel compounds had high efficacy against a collection of MRSA (methicillin resistant S. aureus) clinical isolates recovered from hospitalized or cystic fibrosis patients. The greatest challenge of antibiotic therapy in CF (cystic fibrosis) patients is Staphylococcus aureus (and specifically MRSA) infection because this bacterium’s persistent infection is significantly linked to increasing rates of respiratory function loss and high mortality. As a result, new ways of combating this type of bacterium are required, and they should be based on new antimicrobials, most likely in combination with existing ones [43][44].
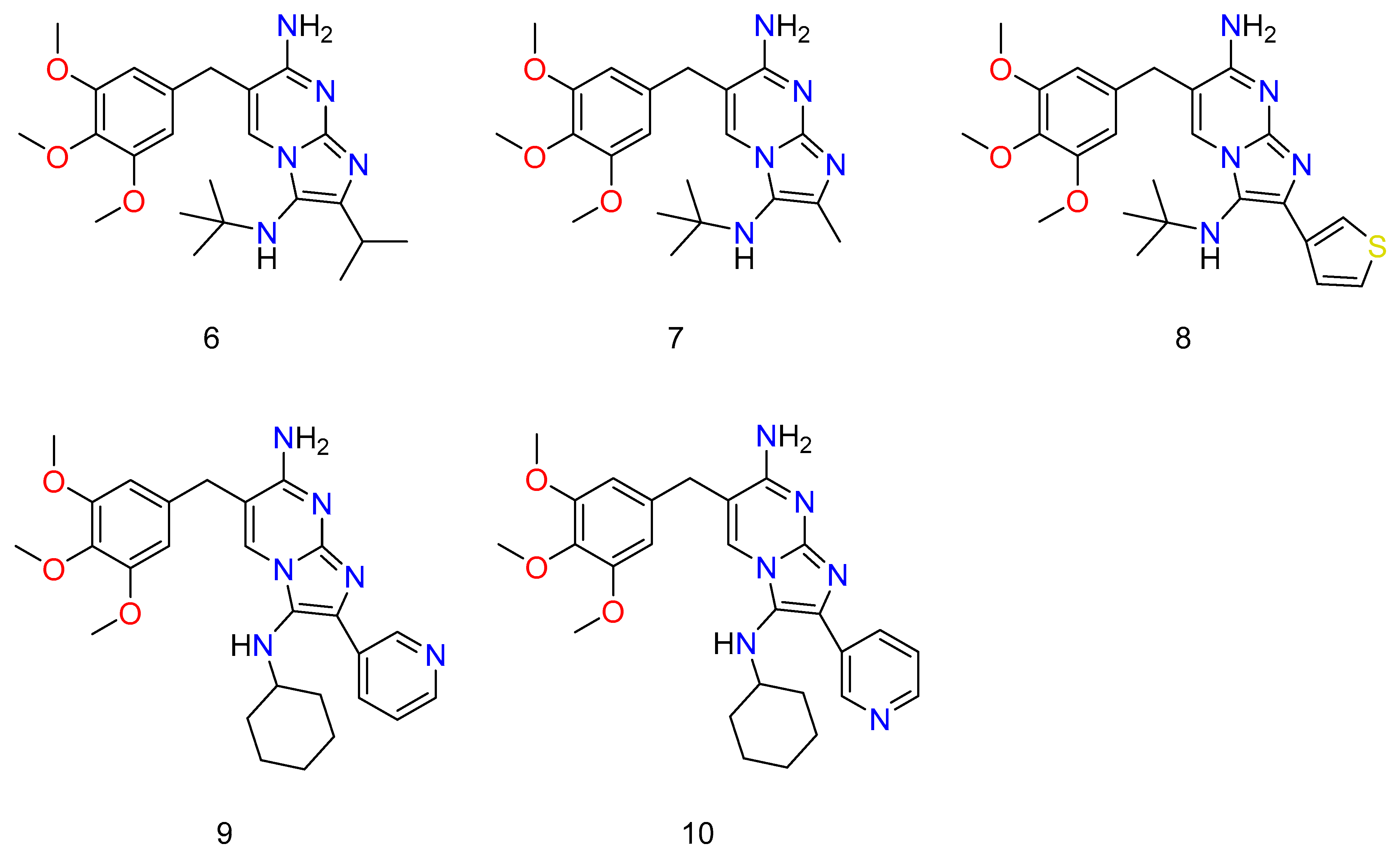
Figure 7. Some novel compounds of TMP, which have potential activity against bacterium. Wherein, 6 is N3-(tert-butyl)-2-isopropyl-6-(3,4,5-trimethoxybenzyl)imidazo[1,2-a]pyrimidine-3,7-diamine; 7 is N3-(tert-butyl)-2-methyl-6-(3,4,5-trimethoxybenzyl)imidazo[1,2-a]pyrimidine-3,7-diamine; 8 is N3-(tert-butyl)-2-(thiophen-3-yl)-6-(3,4,5-trimethoxybenzyl)imidazo [1,2-a]pyrimidine-3,7-diamine; 9 is N3-cyclohexyl-2-(pyridin-3-yl)-6-(3,4,5-trimethoxybenzyl)imidazo[1,2-a]pyrimidine-3,7-diamine; and 10 is N3-cyclohexyl-2-(pyridin-3-yl)-6-(3,4,5-trimethoxybenzyl)imidazo[1,2-a]pyrimidine-3,7-diamine.
Regarding the SMX combination, there was a significant potency (Figure 8: 11–12) (Figure 9: 13–16). Unfortunately, it was found that the adducts, regardless of whether they were alone or in conjunction with SMX, had no effect on Pseudomonas aeruginosa in any circumstance, despite the presence of TMP activity.
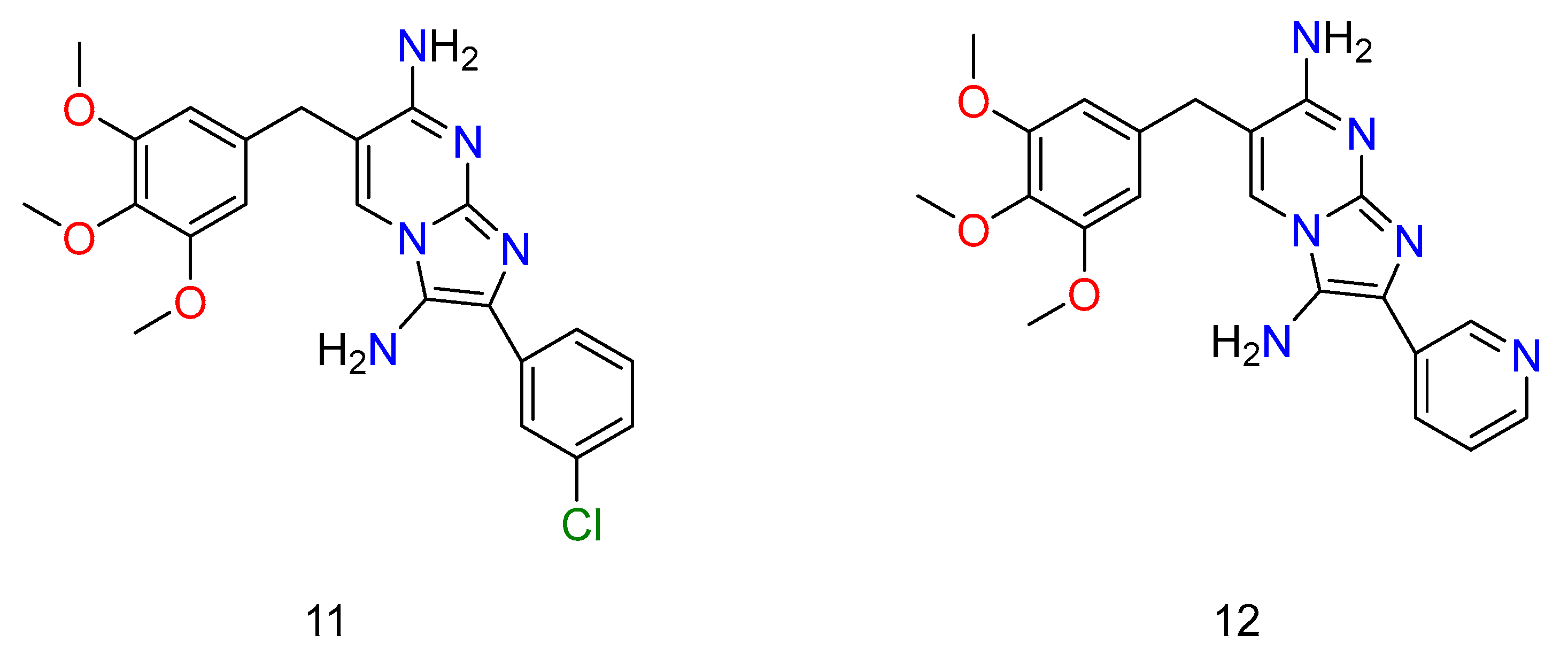
Figure 8. Some of the derivatives of TMP, which have shown potential activity against bacterium. Wherein, 11 is 2-(3-chlorophenyl)-6-(3,4,5-trimethoxybenzyl)imidazo[1,2-a]pyrimidine-3,7-diamine and 12 is 2-(pyridin-3-yl)-6-(3,4,5-trimethoxybenzyl)imidazo[1,2-a]pyrimidine-3,7-diamine.
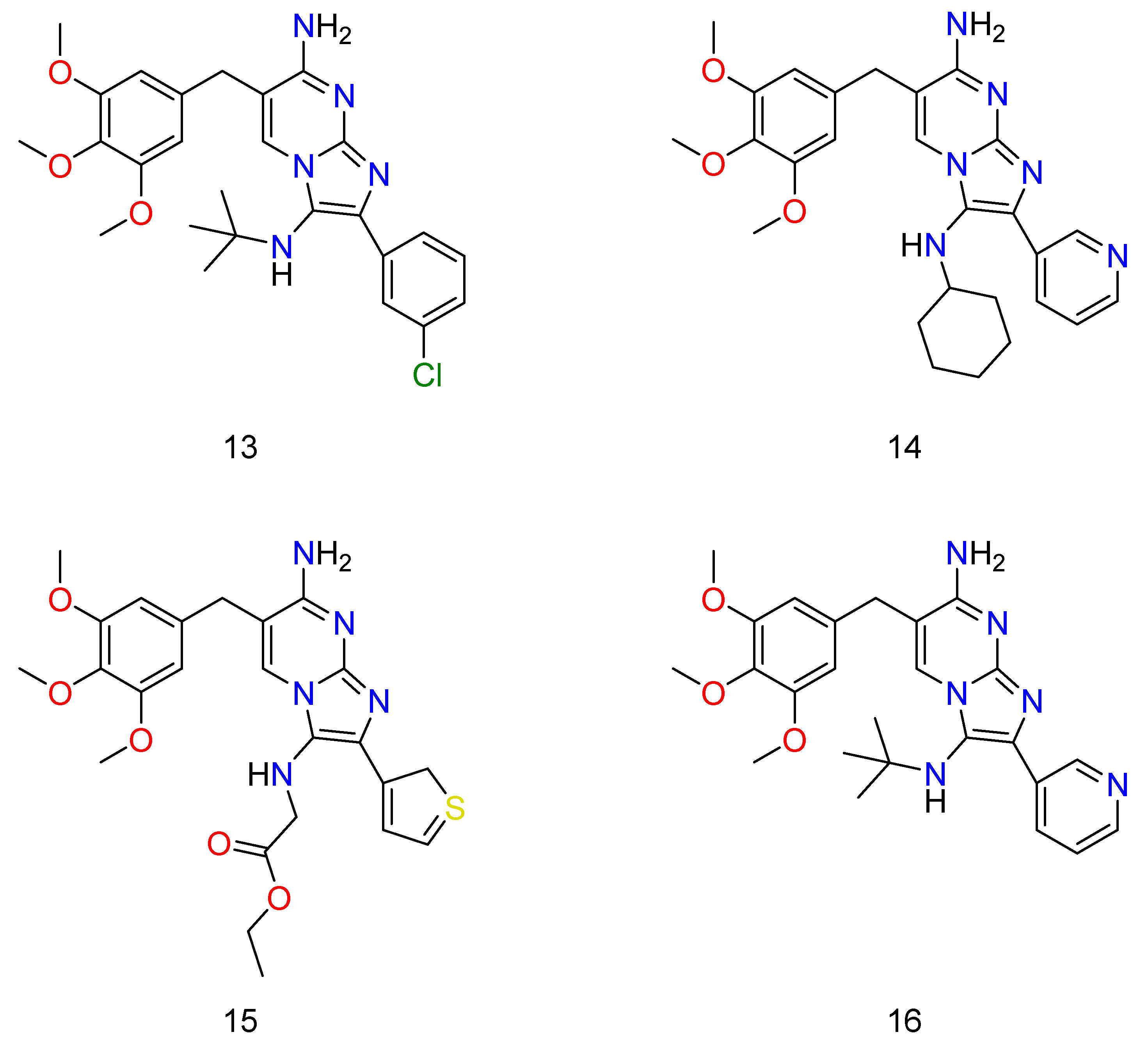
Figure 9. Some of the TMP derivatives that have shown potential activity against bacteria when used in conjunction with SMX. Wherein, 13 is N3-(tert-butyl)-2-(3-chlorophenyl)-6-(3,4,5-trimethoxybenzyl)imidazo[1,2-a]pyrimidine-3,7-diamine; 14 is N3-cyclohexyl-2-(pyridin-3-yl)-6-(3,4,5-trimethoxybenzyl)imidazo[1,2-a]pyrimidine-3,7-diamine; 15 is ethyl (7-amino-2-(2H-1λ3-thiophen-3-yl)-6-(3,4,5-trimethoxybenzyl)imidazo[1,2-a]pyrimidin-3-yl)glycinate; and 16 is N3-(tert-butyl)-2-(pyridin-3-yl)-6-(3,4,5-trimethoxybenzyl)imidazo[1,2-a]pyrimidine-3,7-diamine.
4. The Discovery of Antimicrobial Compounds against Infectious Diseases through the Application of Ugi’s Reaction
Multicomponent reactions have been used by a number of institutions and pharmaceutical businesses to produce medications that aim to combat infectious diseases caused by bacteria, viruses, and parasites. Morphochem created a series of antituberculosis compounds based on the structure of isoniazid and pyrazinamide (Figure 10) [45]. Two libraries of 192 new compounds were created using the pyridine-4- carboxy and pyrazine carboxy pharmacophores present in isoniazid and pyrazinamide as part of the carboxylic acid component in the Ugi reaction. The libraries were made up of individual compounds in 96-well plates, and the raw materials were evaluated against M. tuberculosis after the reaction solvent had evaporated. Compounds that inhibited M. tuberculosis H37Rv by more than 90% were resynthesized and purified, and their minimum inhibitory activity (MIC) and cytotoxicity (IC50) were assessed against the H37Rv strain of M. tuberculosis. The preliminary findings were promising, as numerous compounds from each library had cellular activity similar to isoniazid.
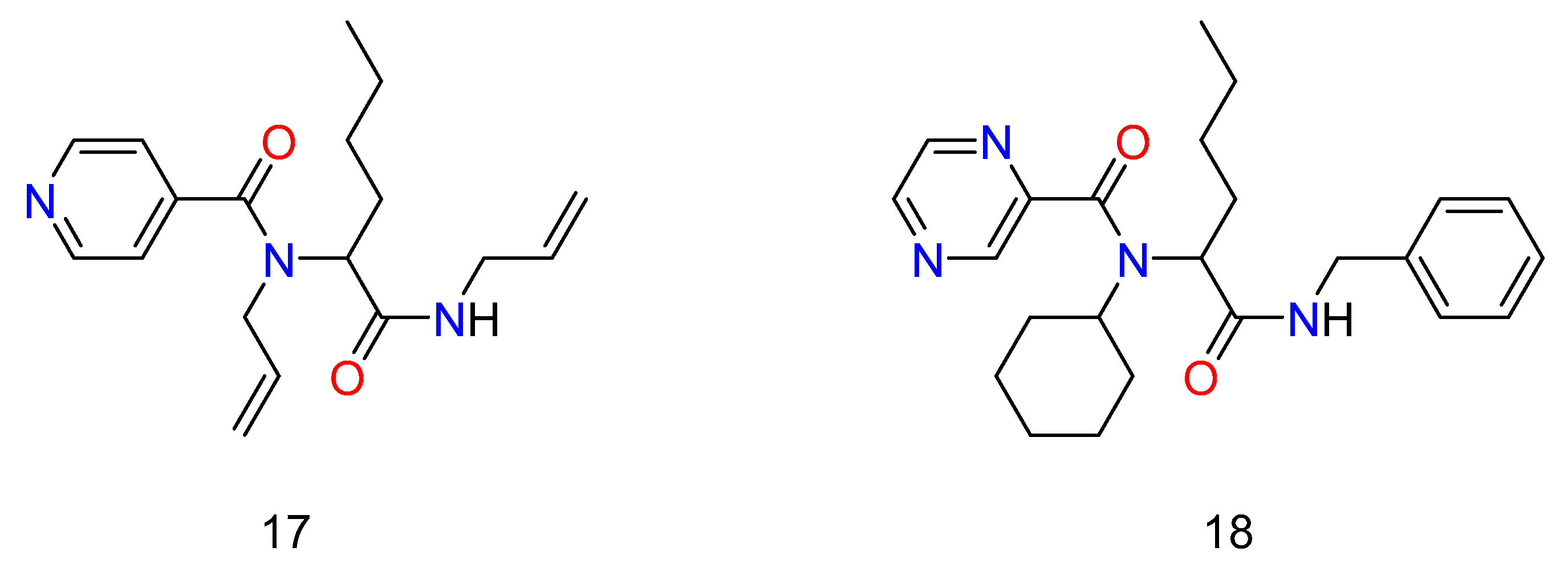
Figure 10. Antituberculosis agents made by the synthesis of Ugi’s Reaction.
The Ugi/Joullie’ reaction [46] was used to make a library of pyrrolidones (Figure 11), which were evaluated against a range of targets and found to be active against the bovine diarrhea virus (BVDV), which is used as a surrogate for the human hepatitis C virus [47]. The compounds were found to be inactive in a variety of glycosidase assays and against the hepatitis B virus, indicating a novel and specific mechanism of action.
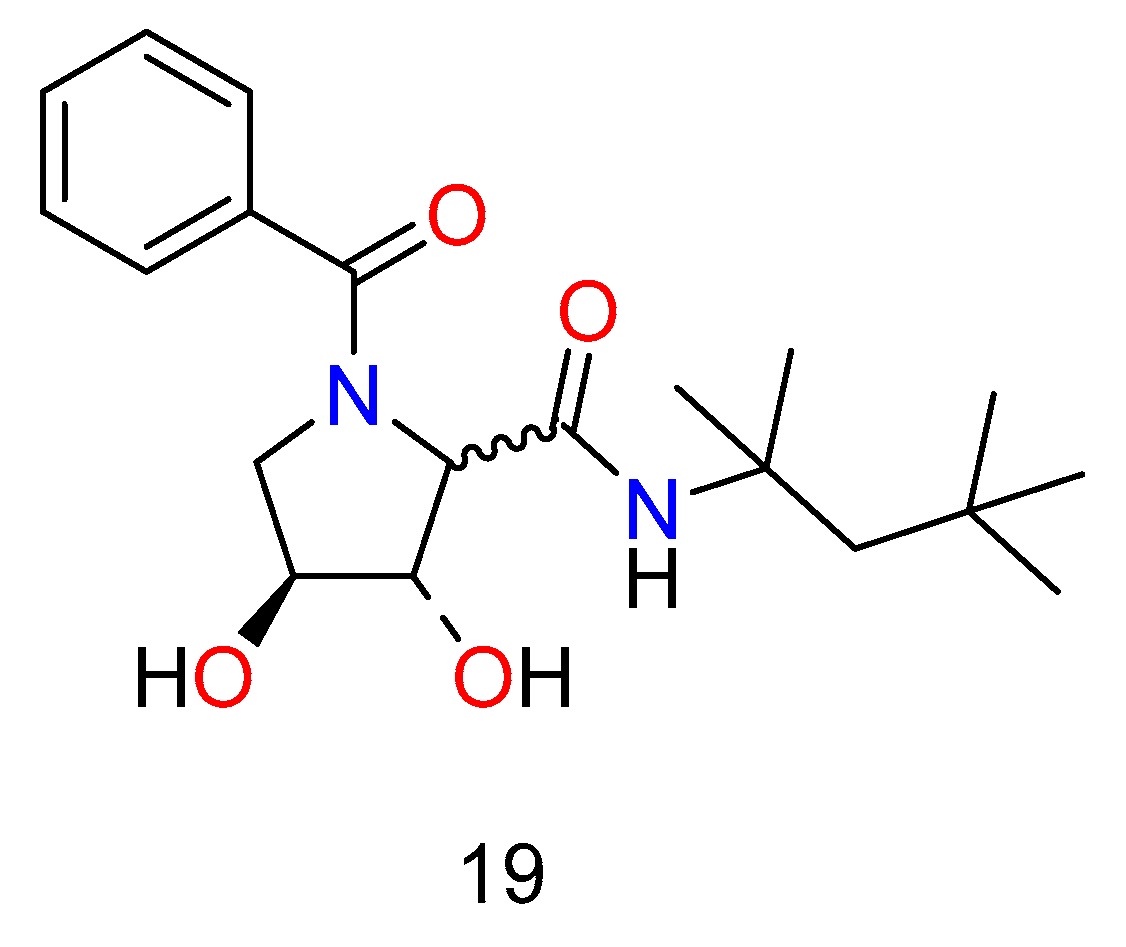
Figure 11. Hepatitis agents synthesized by the Ugi or Joullie reaction.
Early findings on the preparation and testing of a small library of 25 2’-deoxyuridine analogs as antiviral and antileishmanial drugs were reported by Torrence et al. (Figure 12) [48]. The compounds were isolated and evaluated as single diastereomers against cowpox virus (a surrogate for smallpox virus) and the parasite Leishmania donovani, which were produced as diastereomeric mixtures by the Ugi reaction using 5-formyl-2’- deoxyuridine as the aldehyde component. Several compounds, particularly as antileishmanial agents, showed potential.
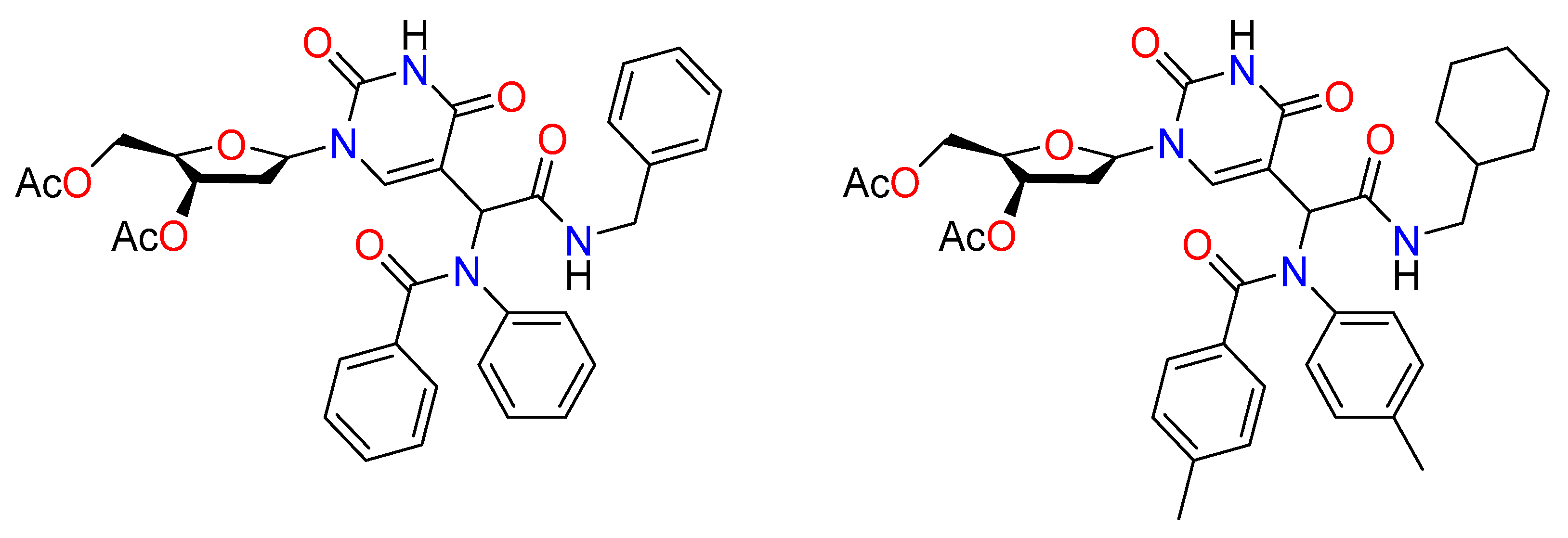
Figure 12. Antiviral agents synthesized by the Ugi reaction.
The quinoline substructure seen in many antimalarial medications was integrated into an amine component for the Ugi reaction using a technique similar to that used in antituberculosis libraries [49]. Despite the fact that several compounds from these libraries had antimalarial activity, the initial libraries’ flexibility and peptidic nature, as well as their low activity, prompted the researchers to pursue a variant of the Ugi reaction that employs two functional groups in one molecule, resulting in heterocyclic structures [50]. A library of 16 4-aminoquinoline γ and δ lactams was synthesized and evaluated against parasite cells of the chloroquine-resistant P. falciparum W2 strain as well as enzymatic activity against recombinant falcipain-2. The compound was found to be more effective against the resistant P. falciparum strain than the antimalarial medication chloroquine [51].
In 1999, a convertible isonitrile was synthesized by a group of researchers led by Linderman et al. Later, this convertible isonitrile was utilized in the process of Ugi reaction to make Uracil Polyoxin, which is also known as analogs of methyl ester (UPOC) [52]. In the field of agriculture, polyoxins are used as fungicides. Polyoxins are a class of nucleoside antibiotics that have a critical natural function in that they oppose chitin synthase (CS). Generally, Polyoxins were isolated from Saccharomyces cerevisiae and Candida albicans due to their antibacterial properties [53]. Chitin synthases are considered attractive targets for inhibition in fungi and insects. Structurally related nikkomycins and polyoxins were synthesized using the Ugi process. In the Ugi process, convertible isonitrile, 2’,3’-isopropylidine-protected uridine-5’-aldehyde, isoxazolecarboxylic acid derivative, and 2,4-dimethoxybenzylamine were included [54].
Later the acidic hydrolysis resulted in the full deprotection of the isopropylidene and DMB groups, and also the isonitrile-extracted amide was transformed into equal UPOC methyl ester or Polyoxins.
A chemical compound named viridic acid was initially isolated from P. viridicatum. This compound is a tetrapeptide that can be produced by numerous Penicillium species. In 2012, Neves et al. employed the Ugi reaction to make a racimate combination of viridic acid. They applied Ugi reaction in the process to shorten the pathway of other standard procedures [55]. The Ugi four-component reaction between compound 1a, 1b, 1c, and the 1d, 1e has given a new compound, 1f, and saponification of 1f caused the racemic viridic acid to reach 83%. Additionally, after traditional separation procedures failed to separate the epimers, combined acid was tested as an antimicrobial drug in the opposition of a bacteria called Aliivibrio fischeri (Gram negative). With the half-maximal inhibitory concentration values of 45.0 ± 4.4 and 38.4 ± 5.8 M, respectively, the compounds were the most powerful [55][56][57][58].
Drug efflux pump inhibitors have a lot of promise as pharmacological treatments for restoring drug sensitivity in multidrug-resistant bacterial infections. The Ugi reaction was utilized to make a targeted collection of C-capped dipeptide efflux pump inhibitors, with C-capped dipeptides BU-005 being made using the Ugi four-compound reaction and full deprotection of Boc and DMB of the product using TFA. The C-capped dipeptide BU-005 was able to inhibit two chloramphenicol-specific efflux pumps in Streptomyces coelicolor, a Gram-positive bacterium that is related to the human pathogen Mycobacterium TB [59][60].
A member of uridylpeptide antibiotics named Pacidamycin D 90 was initially discovered in 1989 from Streptomyces coeruleorubidus AB 1183F-64. 3′-hydroxypacidamycin D are analogs of Pacidamycin D 90 which are also considered to be uridylpeptide antibiotics that are selective of antimicrobial drugs against Pseudomonas aeruginosa. They also have the capability to act on the inhibition of phospho-MurNAc-pentapeptide transferase (MraY). The half-maximal inhibitory concentration value is 42 nM, and their MIC for different strains of P. aeruginosa is 8–32 μg mL. MraY is an essential enzyme in bacteria, wherein it is responsible for the creation of lipid I in the peptidoglycan biosynthesis pathway [61][62].
Benzimidazoles have many anti-medicinal properties, such as anti-inflammatory, antibacterial, anti-HIV, and anticancer properties. With the intent of synthesizing a variety of benzimidazole, in 2016, Yan et al. led his research team and synthesized several benzimidazole derivatives. They extended their study to Ugi and aza-Wittig to extract more useful heterocycles than before. Therefore, the scientist team developed a one-pot reaction that combines the reaction of Ugi with a catalytic reaction of aza-Wittig to synthesize multi-substituted benzimidazoles under the presence of a catalytic amount of 2-aminobenzoylazide derivatives, 3-methyl-1-phenyl-2-phospholene, 1-oxide, carboxylic, aldehydes, and isonitriles. The one-pot reaction was designed to produce more poly-substituted benzimidazoles [63][64][65][66][67].
The utility of antimicrobial hydrogels as wound dressings and fillers make them very enticing materials. These types of gels have a lot of water in them, so they keep the wound area moist and well-hydrated, which helps the immune cells work, becoming an essential part of the healing process. Hydrogels that can self-heal are distinct from regular hydrogels since they can automatically fix internal or exterior damage without help. Self-healing hydrogels, a new class of intelligent soft matter, have been the subject of extensive studies and demonstrated remarkable potential as biomaterials for medication delivery and treatment. In 2019, Zeng and his research team developed a self-healing antibacterial hydrogen with the application of Ugi’s reaction [68]. They achieved a multifunctional polyethylene glycol through UGi’s reaction by efficiently linking between phenols groups and phenylboronic acid at the end of a polyethylene glycol derivative. In pre-clinical trials, the multifunctional polyethylene glycol had shown antibacterial properties primarily due to the phenol moieties present in the polymer structure. When the multifunctional polyethylene glycol was mixed with polyvinyl alcohol, it crosslinked the polyethylene glycol through dynamic borate esters between phenylboronic acid moieties in itself and diol groups in polyvinyl alcohol [68]. Thus, this is how they have produced self-healing antibacterial hydrogels, which can have fifty- and ten-times higher MIC values than the combination of penicillin and streptomycin for E. coli and or S. aureus, respectively.
This entry is adapted from the peer-reviewed paper 10.3390/antibiotics12050849
References
- Zarganes-Tzitzikas, T.; Chandgude, A.L.; Dömling, A. Multicomponent reactions, union of MCRs and beyond. Chem. Rec. 2015, 15, 981–996.
- Afshari, R.; Shaabani, A. Materials functionalization with multicomponent reactions: State of the art. ACS Comb. Sci. 2018, 20, 499–528.
- Quazi, S.; Gavas, S.; Malik, J.A.; Suman, K.S.; Haider, Z. In-silico pharmacophore and molecular docking based drug discovery against marburg virus’s viral protein 35; A potent of MAVD. bioRxiv 2021. preprint.
- Lieke, W. Ueber das cyanallyl. Justus Liebigs Ann. Chem. 1859, 316–321.
- Gautier, A. Ueber die einwirkung des chlorwasserstoffs u. a. Auf das aethyl- und methylcyanür. Ann. Chem. Pharm. 1867, 142, 289–294.
- Ugi, I. The α-addition of immonium ions and anions to isonitriles accompanied by secondary reactions. Angew. Chem. Int. Ed. Engl. 1962, 1, 8–21.
- Ugi, I.; Dömling, A.; Gruber, B.; Almstetter, M. Multicomponent reactions and their libraries—A new approach to preparative organic chemistry. Croat. Chem. Acta 1997, 70, 631–647.
- Ugi, I. Neuere methoden der präparativen organischen chemie IV: Mit sekundär-reaktionen gekoppelte α-additionen von immonium-ionen und anionen an isonitrile. Angew. Chem. 1962, 74, 9–22.
- Ugi, I.; Meyr, R.; Fetzer, U.; Steinbrückner, C. Versuche mit isonitrilen. Angew. Chem. 1959, 71, 386.
- Arshady, R.; Ugi, I. Solid phase peptide synthesis by four component condensation: Peptide formation on an isocyano polymer support. Z. Naturforsch. B J. Chem. Sci. 1981, 36, 1202–1203.
- Oertel, K.; Zech, G.; Kunz, H. Stereoselective combinatorial ugi-multicomponent synthesis on solid phase this work was supported by the deutsche forschungsgemeinschaft and by the fonds der chemischen industrie. Angew. Chem. Int. Ed. Engl. 2000, 39, 1431–1433.
- Bienaymé, H.; Bouzid, K. A new heterocyclic multicomponent reaction for the combinatorial synthesis of fused 3-aminoimidazoles. Angew. Chem. Int. Ed 1998, 37, 2234–2237.
- Blackburn, C.; Guan, B.; Fleming, P.; Shiosaki, K.; Tsai, S. Parallel synthesis of 3-aminoimidazo pyridines and pyrazines by a new three-component condensation. Tetrahedron Lett. 1998, 39, 3635–3638.
- Groebke, K.; Weber, L.; Mehlin, F. ChemInform abstract: Synthesis of imidazo annulated pyridines, pyrazines, and pyrimidines by a novel three-component condensation. ChemInform 2010, 29.
- Ugi, I.; Meyr, R.; Isonitrile, V. Erweiterter anwendungsbereich der passerini-reaktion. Chem. Ber. 1961, 94, 2229–2233.
- Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Catalytic enantioselective passerini three-component reaction. Angew. Chem. Int. Ed. Engl. 2008, 47, 388–391.
- Brioche, J.; Masson, G.; Zhu, J. Passerini three-component reaction of alcohols under catalytic aerobic oxidative conditions. Org. Lett. 2010, 12, 1432–1435.
- Yanai, H.; Oguchi, T.; Taguchi, T. Direct alkylative passerini reaction of aldehydes, isocyanides, and free aliphatic alcohols catalyzed by indium (III) triflate. J. Org. Chem. 2009, 74, 3927–3929.
- El Kaim, L.; Gizolme, M.; Grimaud, L. O-arylative passerini reactions. Org. Lett. 2006, 8, 5021–5023.
- Soeta, T.; Kojima, Y.; Ukaji, Y.; Inomata, K. O-silylative passerini reaction: A new one-pot synthesis of α-siloxyamides. Org. Lett. 2010, 12, 4341–4343.
- Sunderhaus, J.D.; Martin, S.F. Applications of multicomponent reactions to the synthesis of diverse heterocyclic scaffolds. Chemistry 2009, 15, 1300–1308.
- Paulvannan, K. Preparation of tricyclic nitrogen heterocycles via tandem four-component condensation/intramolecular diels-alder reaction. Tetrahedron Lett. 1999, 40, 1851–1854.
- Koopmanschap, G.; Ruijter, E.; Orru, R.V.A. Isocyanide-based multicomponent reactions towards cyclic constrained peptidomimetics. Beilstein J. Org. Chem. 2014, 10, 544–598.
- Bonnaterre, F.; Bois-Choussy, M.; Zhu, J. Rapid access to oxindoles by the combined use of an ugi four-component reaction and a microwave-assisted intramolecular buchwald−hartwig amidation reaction. Org. Lett. 2006, 8, 4351–4354.
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524.
- Quazi, S.; Jangi, R. Artificial intelligence and machine learning in medicinal chemistry and validation of emerging drug targets. Adv. Control. Drug Deliv. Syst. 2021, 27–43. Available online: https://www.igi-global.com/chapter/artificial-intelligence-and-machine-learning-in-medicinal-chemistry-and-validation-of-emerging-drug-targets/300400 (accessed on 1 March 2023).
- Hulme, C.; Lee, Y.-S. Emerging approaches for the syntheses of bicyclic imidazo -heterocycles. Mol. Divers. 2008, 12, 1–15.
- Devi, N.; Rawal, R.K.; Singh, V. Diversity-oriented synthesis of fused-imidazole derivatives via groebke–blackburn–bienayme reaction: A review. Tetrahedron 2015, 71, 183–232.
- Akritopoulou-Zanze, I.; Wakefield, B.D.; Gasiecki, A.; Kalvin, D.; Johnson, E.F.; Kovar, P.; Djuric, S.W. Scaffold oriented synthesis. Part 4: Design, synthesis and biological evaluation of novel 5-substituted indazoles as potent and selective kinase inhibitors employing heterocycle forming and multicomponent reactions. Bioorg. Med. Chem. Lett. 2011, 21, 1480–1483.
- Baviskar, A.T.; Madaan, C.; Preet, R.; Mohapatra, P.; Jain, V.; Agarwal, A.; Guchhait, S.K.; Kundu, C.N.; Banerjee, U.C.; Bharatam, P.V. N-fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase IIα and induce apoptosis in G1/S phase. J. Med. Chem. 2011, 54, 5013–5030.
- Shukla, N.M.; Salunke, D.B.; Yoo, E.; Mutz, C.A.; Balakrishna, R.; David, S.A. Antibacterial activities of groebke–blackburn–bienaymé-derived imidazo pyridin-3-amines. Bioorg. Med. Chem. 2012, 20, 5850–5863.
- Burchak, O.N.; Mugherli, L.; Ostuni, M.; Lacapère, J.J.; Balakirev, M.Y. Combinatorial discovery of fluorescent pharmacophores by multicomponent reactions in droplet arrays. J. Am. Chem. Soc. 2011, 133, 10058–10061.
- Elleder, D.; Baiga, T.J.; Russell, R.L.; Naughton, J.A.; Hughes, S.H.; Noel, J.P.; Young, J.A.T. Identification of a 3-aminoimidazo pyridine inhibitor of HIV-1 reverse transcriptase. Virol. J. 2012, 9, 305.
- Bode, M.L.; Gravestock, D.; Moleele, S.S.; van der Westhuyzen, C.W.; Pelly, S.C.; Steenkamp, P.A.; Hoppe, H.C.; Khan, T.; Nkabinde, L.A. Imidazo pyridin-3-amines as potential HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. 2011, 19, 4227–4237.
- Urbancic, K.F.; Ierino, F.; Phillips, E.; Mount, P.F.; Mahony, A.; Trubiano, J.A. Taking the challenge: A protocolized approach to optimize pneumocystis pneumonia prophylaxis in renal transplant recipients. Am. J. Transpl. 2018, 18, 462–466.
- Heaslet, H.; Harris, M.; Fahnoe, K.; Sarver, R.; Putz, H.; Chang, J.; Subramanyam, C.; Barreiro, G.; Miller, J.R. Structural comparison of chromosomal and exogenous dihydrofolate reductase from staphylococcus aureus in complex with the potent inhibitor trimethoprim. Proteins 2009, 76, 706–717.
- Zhou, W.; Scocchera, E.W.; Wright, D.L.; Anderson, A.C. Antifolates as effective antimicrobial agents: New generations of trimethoprim analogs. Medchemcomm 2013, 4, 908.
- Quazi, S.; Malik, J.; Suman, K.S.; Capuzzo, A.M.; Haider, Z. Discovery of potential drug-like compounds against viral protein (VP40) of marburg virus using pharmacophoric based virtual screening from ZINC database. bioRxiv 2021.
- Lombardo, M.N.; G-Dayanandan, N.; Wright, D.L.; Anderson, A.C. Crystal structures of trimethoprim-resistant DfrA1 rationalize potent inhibition by propargyl-linked antifolates. ACS Infect. Dis. 2016, 2, 149–156.
- Rashid, U.; Ahmad, W.; Hassan, S.F.; Qureshi, N.A.; Niaz, B.; Muhammad, B.; Imdad, S.; Sajid, M. Design, synthesis, antibacterial activity and docking study of some new trimethoprim derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 5749–5753.
- Pedrola, M.; Jorba, M.; Jardas, E.; Jardi, F.; Ghashghaei, O.; Viñas, M.; Lavilla, R. Multicomponent reactions upon the known drug trimethoprim as a source of novel antimicrobial agents. Front. Chem. 2019, 7, 475.
- Ghashghaei, O.; Caputo, S.; Sintes, M.; Revés, M.; Kielland, N.; Estarellas, C.; Luque, F.J.; Aviñó, A.; Eritja, R.; Serna-Gallego, A.; et al. Multiple multicomponent reactions: Unexplored substrates, selective processes, and versatile chemotypes in biomedicine. Chemistry 2018, 24, 14513–14521.
- Dolce, D.; Neri, S.; Grisotto, L.; Campana, S.; Ravenni, N.; Miselli, F.; Camera, E.; Zavataro, L.; Braggion, C.; Fiscarelli, E.V.; et al. Methicillin-resistant staphylococcus aureus eradication in cystic fibrosis patients: A randomized multicenter study. PLoS ONE 2019, 14, e0213497.
- Lo, D.K.; Muhlebach, M.S.; Smyth, A.R. Interventions for the eradication of meticillin-resistant staphylococcus aureus (MRSA) in people with cystic fibrosis. Cochrane Database Syst. Rev. 2018, 7, CD009650.
- Xhemali, X.; Smith, J.R.; Kebriaei, R.; Rice, S.A.; Stamper, K.C.; Compton, M.; Singh, N.B.; Jahanbakhsh, S.; Rybak, M.J. Evaluation of dalbavancin alone and in combination with β-lactam antibiotics against resistant phenotypes of staphylococcus aureus. J. Antimicrob. Chemother. 2019, 74, 82–86.
- Dömling, A.; Achatz, S.; Beck, B. Novel anti-tuberculosis agents from MCR libraries. Bioorg. Med. Chem. Lett. 2007, 17, 5483–5486.
- Nutt, R.F.; Joullie, M.M. Four-component condensation: A new versatile method for the synthesis of substituted prolyl peptides. J. Am. Chem. Soc. 1982, 104, 5852–5853.
- Akritopoulou-Zanze, I. Isocyanide-based multicomponent reactions in drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 324–331.
- Fan, X.; Zhang, X.; Bories, C.; Loiseau, P.M.; Torrence, P.F. The ugi reaction in the generation of new nucleosides as potential antiviral and antileishmanial agents. Bioorg. Chem. 2007, 35, 121–136.
- Musonda, C.C.; Taylor, D.; Lehman, J.; Gut, J.; Rosenthal, P.J.; Chibale, K. Application of multicomponent reactions to antimalarial drug discovery. Part 1. Parallel synthesis and antiplasmodial activity of new 4-aminoquinoline ugi adducts. Bioorg. Med. Chem. Lett. 2004, 14, 3901–3905.
- Short, K.M.; Ching, B.W.; Mjalli, A.M.M. Exploitation of the ugi 4CC reaction: Preparation of small molecule combinatorial libraries via solid phase. Tetrahedron 1997, 53, 6653–6679.
- Musonda, C.C.; Gut, J.; Rosenthal, P.J.; De Souza, C.; Chibale, R.C. Application of multicomponent reactions to antimalarial drug discovery. Part 2. New antiplasmodial and antitrypanosomal 4-aminoquinoline g-and d-lactams via a ‘catch and release’ protocol. Bioorg. Med. Chem. 2006, 14, 5605–5615.
- Linderman, R.J.; Binet, S.; Petrich, S.R. Enhanced diastereoselectivity in the asymmetric ugi reaction using a new “convertible” isonitrile. J. Org. Chem. 1999, 64, 8058.
- Cohen, E. Chitin biochemistry: Synthesis and inhibition. Annu. Rev. Entomol. 1987, 32, 71–93.
- Plant, A.; Thompson, P.; Williams, D.M. Application of the ugi reaction for the one-pot synthesis of uracil polyoxin C analogues. J. Org. Chem. 2009, 74, 4870–4873.
- Neves Filho, R.A.W.; Stark, S.; Westermann, B.; Wessjohann, L.A. The multicomponent approach to N-methyl peptides: Total synthesis of antibacterial (−)-viridic acid and analogues. Beilstein J. Org. Chem. 2012, 8, 2085–2090.
- Brase, S.; Encinas, A.; Keck, J.; Nising, C.F. Chemistry and biology of mycotoxins and related fungal metabolites. Chem. Rev. 2009, 109, 3903–3990.
- Quazi, S. Role of artificial intelligence and machine learning in bioinformatics: Drug discovery and drug repurposing. Preprints 2021, 2021050346.
- Iarani, G.M.; Moradi, R.; Mahammadkhani, L. Application of multicomponent reactions in the total synthesis of natural peptides. Org. Chem. 2019, 18–40.
- Okandeji, B.O.; Greenwald, D.M.; Wroten, J.; Sello, J.K. Synthesis and evaluation of inhibitors of bacterial drug efflux pumps of the major facilitator superfamily. Bioorg. Med. Chem. 2011, 19, 7679–7689.
- Okamoto, K.; Sakagami, M.; Feng, F.; Togame, H.; Takemoto, H.; Ichikawa, S.; Matsuda, A. Total synthesis of pacidamycin D by Cu (I)-catalyzed oxy enamide formation. Org. Lett. 2011, 13, 5240–5243.
- Winn, M.; Goss, R.J.; Kimura, K.I.; Bugg, T.D. Antimicrobial nucleoside antibiotics targeting cell wall assembly: Recent advances in structure–function studies and nucleoside biosynthesis. Nat. Prod. Rep. 2010, 27, 279–304.
- Fernandes, P.B.; Swanson, R.N.; Hardy, D.J.; Hanson, C.W.; Coen, L.; Rasmussen, R.R.; Chen, R.H. Pacidamycins, a novel series of antibiotics with anti-pseudomonas aeruginosa activity. III. Microbiologic profile. J. Antibiot. 1989, 42, 521–526.
- Yan, Y.-M.; Rao, Y.; Ding, M.-W. One-pot synthesis of multisubstituted benzimidazoles via sequential ugi and catalytic aza-wittig reaction starting from 2-aminobenzoyl azides. J. Org. Chem. 2016, 81, 1263–1268.
- Mavrova, A.T.; Yancheva, D.; Anastassova, N.; Anichina, K.; Zvezdanovic, J.; Djordjevic, A.; Markovic, D.; Smelcerovic, A. Synthesis, electronic properties, antioxidant and antibacterial activity of some new benzimidazoles. Bioorg. Med. Chem. 2015, 23, 6317–6326.
- Pan, T.; He, X.; Chen, B.; Chen, H.; Geng, G.; Luo, H.; Zhang, H.; Bai, C. Development of benzimidazole derivatives to inhibit HIV-1 replication through protecting APOBEC3G protein. Eur. J. Med. Chem. 2015, 95, 500–513.
- Vasantha, K.; Basavarajaswamy, G.; Rai, M.V.; Boja, P.; Pai, V.R.; Shruthi, N.; Bhat, M. Rapid ‘one-pot’synthesis of a novel benzimidazole-5-carboxylate and its hydrazone derivatives as potential anti-inflammatory and antimicrobial agents. Bioorg. Med. Chem. Lett. 2015, 25, 1420–1426.
- Zeng, Y.; Li, Y.; Liu, G.; Wei, Y.; Wu, Y.; Tao, L. Antibacterial self-healing hydrogel via the ugi reaction. ACS Appl. Polym. Mater. 2019, 2, 404–410.
This entry is offline, you can click here to edit this entry!