1. Role of cGAS–STING Pathway in Alzheimer’s Disease
One of the key pathophysiological mechanisms implicated in AD is mitochondrial dysfunction, characterized by mitochondrial damage and dysfunctional mitophagy caused by cellular stress due to the accumulation of toxic protein aggregates such as amyloid-β or hyperphosphorylated tau [
57,
58,
59]. These proteins induce oxidative damage of mtDNA and induce double-stranded breaks in DNA [
60]. Hence, the release of mtDNA and dsDNA fragments into the cytosol can act as a ligand for cGAS, which further activates STING and promotes a neuroinflammatory response [
61]. Interestingly, the cGAS is negatively regulated by apoptosis in that the release of DNA from apoptotic cells does not activate the cGAS–STING pathway. However, it is activated by DNA released from intact cells undergoing cellular stress [
62].
In post-mortem human AD brain samples, increased STING expression was noted predominantly in CNS microvasculature and neuronal cells compared to age-matched control brain samples. The increased STING levels in neuronal cells were in close proximity to the amyloid plaques, and this colocalization of Aβ near STING expression may be due to the endoplasmic reticulum and mitochondrial stress on the cells caused by toxic protein accumulation, as previously indicated. Interestingly, STING expression was less evident in GFAP-positive astrocytes and CD68-positive microglia in the AD tissue [
64]. Similarly, in a 5xFAD mouse model of AD, the activation of STING–IFN signaling following cGAS–dsDNA interaction was present mainly in the microglia but not in the neurons or astrocytes [
63]. A study by Hou et al., demonstrated increased expression of cGAS and STING proteins in the brains of APP/PS1 mice in comparison to wild-type mice. Furthermore, treatment of HMC3 human microglial cells with the STING inhibitor H-151 prevented Aβ42-induced IL-6 production. Since the downstream signaling of STING activation involves a type I IFN response, and elevated levels of type I IFNs have been found in post-mortem human AD brains [
65], the activation of STING could be linked to AD progression.
Neuroinflammation is considered to be a critical component in the pathogenesis of AD [
66]. Accumulation of Aβ plaques, neurofibrillary tangles and degenerating neurons are classical stimulators of AD neuroinflammation [
67,
68]. Several lines of evidence indicate that cGAS and STING are predominantly expressed in the microglia, and the proinflammatory responses induced by activation of the cGAS–STING pathway are potent in microglia but less so in neurons and astrocytes [
69,
70]. In line with this, in a mouse model of chronic neurodegeneration, the upregulation of cytosolic DNA sensors, including cGAS, were found to drive type 1 IFN and pro-inflammatory cytokine production in microglia [
71]. Similarly, the microglial cGAS–STING pathway has been shown to play a crucial role in mediating inflammation due to tau pathology [
72]. In the 5xFAD mouse model of AD, the expression of phosphorylated STING co-localized with activated microglial marker CD68 around Aβ plaques suggested that the STING–IFN response occurs mainly in the microglia [
63]. In a high-fat diet obese mouse model for prediabetes and cognitive impairment, increased expression of cGAS and STING was noted in the hippocampus as early as four days following high-fat diet feeding. The acute high-fat diet (HFD) also showed microglial activation and an increase in pro-inflammatory responses without any changes in cognition, indicating that the HFD promotes an acute and early pro-inflammatory response in the CNS that precedes or initiates several signaling cascades leading to neurodegeneration and cognitive impairment with chronic HFD [
73]. Since inflammatory microglia play a critical role in AD pathogenesis and related neuroinflammation [
74,
75], the activation of cGAS–STING and its type I IFN respons, which releases various cytokines, can structurally and functionally injure the neurons [
76].
The inflammatory response is one of the hallmarks of cellular senescence and SASPs are documented in various mouse models of AD [
77,
78]. As described earlier, the cGAS–STING pathway is linked to cellular senescence and inflammation. In a mouse model of AD (APP/PS1), cGAS expression was increased in 7-, 12- and 20-month-old AD mice in comparison to wild type. Furthermore, STING expression was higher in AD mice only after 12 months. The differences in cGAS and STING activation at different age groups could be attributed to different levels of immune activation at different ages. Furthermore, markers of cellular senescence (SA-β-gal staining and p16INK4a) increased in the hippocampus and cortex of AD mice. A microarray analysis on the hippocampus and cortex from 12-month-old APP/PS1 AD mice showed several immune and inflammatory response genes to be upregulated. The NOD, LRR and pyrin domain-containing protein 3 (NLRP3) inflammasome is a cytosolic signaling complex that is mainly expressed in microglia and plays a role in their inflammatory responses [
79]. Increased expression of NLRP3, caspase-1 and NF-κB along with positive immunostaining for IBA1 (marker of microglial activation) and GFAP (marker of astrocyte reactivity) were noted in the cortex of APP/PS1 mice. Several lines of evidence indicated that cGAS–STING detects cytosolic DNA and is activated by DNA damage and neuroinflammation [
34,
80]. γ-H2AX (marker for DNA damage) and cleaved caspase-3 (marker for apoptosis) showed a significant increase in the hippocampus and cortex of AD mouse brains. All these pathological changes were reversed by administering NAD+ precursor nicotinamide riboside (NR) [
81]. Hence, compromised autophagy/mitophagy can lead to increased DNA release in the cytosol, thereby causing cGAS–STING activation, which further leads to neuroinflammation and cellular senescence.
One of the major functions of microglia is to maintain tissue homeostasis by engulfment and clearance of debris. The triggering receptor expressed on myeloid cells 2 (TREM2) is a cell surface receptor on the microglia, which plays a crucial role in the phagocytosis of various substrates such as Aβ, apoptotic neurons and bacteria [
82,
83,
84]. Hence, TREM2 promotes clearance of Aβ and attenuates neuroinflammation by inhibiting the production of pro-inflammatory cytokines [
85,
86,
87]. Furthermore, the anti-inflammatory effect of TREM2 is mediated by a change in microglial polarization from M1 (pro-inflammatory) to M2 (anti-inflammatory phenotype). Administration of recombinant cGAMP in a 5-month APP/PS1 mouse model of AD-restored memory functions, decreased Aβ plaques and neuronal death and ameliorated neuroinflammation. The stimulation of the cGAS–STING pathway induced the expression of triggering receptors expressed on myeloid cells 2 (TREM2), which decreased Aβ and neuronal loss. Furthermore, TREM2 induced microglia polarization from M1 towards M2 phenotype, thereby attenuating neuroinflammatory responses [
88]. Lysophosphatidic acids, an oxidized form of low-density lipoproteins (LDLs), are bioactive signaling phospholipids that are implicated in the pathogenesis of AD [
89]. Aberrant activation of LPA signaling has been shown to increase Aβ levels, phosphorylation of tau and neuritic retraction [
90,
91]. Lysophosphatidic acid treatment in BV2 murine microglial cells increased the protein expression of cGAS and STING. Furthermore, STING induced increased nuclear translocation of NFκB and IRF3 and transcription of several inflammatory genes. The results from these limited studies indicated that microglia play a crucial role in regulating cGAS–STING signaling and neuroinflammatory responses in AD.
There are only limited studies that have evaluated the association between tau pathology and the cGAS–STING pathway. A recent study by Jin et al. showed that the tau protein interacts with Polyglutamine-binding protein 1 (PQBP1) and induces a neuroinflammatory response by activating the cGAS–STING pathway in microglial cells. Microglia-specific depletion of PQBP1 in primary cultures and PQPB1 knockout mice showed that PQPB1 is essential for sensing tau to induce nuclear translocation of NFκB and IRF3 (downstream transcription factors of the cGAS–STING pathway), increased transcription of inflammatory genes such as
Tnf and Isg54. Finally, PQPB1 knockout mice injected with extracellular tau monomer showed less cognitive impairment. These results indicated that PQBP1 acted as an intracellular receptor for tau and activated the cGAS–STING pathway, specifically in the microglial cells [
73]. The abnormal accumulation of tau protein has been shown to induce TE transcription as described earlier [
92], and this unchecked TE expression leads to activation of cGAS–STING pathway, which leads to the activation of interferon regulatory factor 3 (IRF3), induction of type I interferons and activation of NF-κB [
93]. More studies need to be performed to investigate how cGAS–STING regulates tau pathology in AD to determine therapeutic targets. The various mechanisms by which cGAS–STING pathway is involved in AD is summarized in
Figure 2.
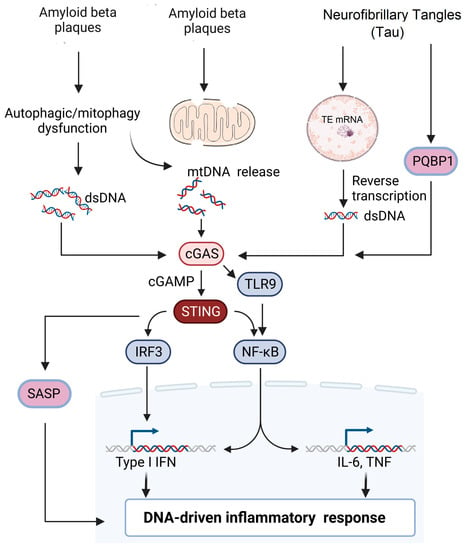
Figure 2. Potential signaling pathways involved in AD pathogenesis. Extracellular accumulation of β-amyloid plaques leads to either mitochondrial damage or impaired mitophagy leading to release of mitochondrial DNA into the cytosol. Impaired autophagy and cellular stress lead to accumulation of cytoplasmic dsDNA leading to the activation of the cGAS–STING pathway. Neurofibrillary tangles (NFT) can induce reverse transcription of transposable elements in the nucleus, leading to the accumulation of dsDNA. Alternatively, PGBP1 acts as an intracellular receptor for tau and activates the cGAS–STING pathway, which subsequently activates either the IRF3 or NF-κB pathway, leading to DNA-driven inflammatory response.
2. Current Therapeutics to Target STING in AD
Overstimulation of the cGAS–STING pathway and its associated neuroinflammation is thought to be associated with the development or progression of various neurodegenerative diseases, including AD. Hence, therapeutics inhibiting this pathway could attenuate disease progression in AD and other neurodegenerative diseases. The goal of developing agents that will inhibit the cGAS–STING pathway is possibly to protect neurons by alleviating chronic inflammation due to mitochondrial stress and autophagic dysregulation. On the other hand, inhibition of this pathway could increase susceptibility to infection that is secondary to the suppression of an innate immune response. Furthermore, the promotion of cancer can occur by limiting cellular senescence in cells with genomic instability/DNA damage. Although there are currently no cGAS–STING inhibitors currently approved for AD, studies using cell culture models and animal studies indicate that certain compounds are beneficial for attenuating the pathologies of AD (Table 1).
Table 1. In vitro and in vivo studies evaluating cGAS–STING inhibitors in Alzheimer’s disease.
Before evaluating STING-based therapies in neuroinflammatory diseases like AD, several considerations must be addressed. First, there is not yet a consensus on which types of brain cell express cGAS and STING. Several studies have demonstrated conflicting evidence regarding the expression of cGAS and STING in different cell types [
94,
95,
96]. For instance, activation of the STING–IFN cascade by cGAS–dsDNA interactions has been shown to mainly occur in the microglia and not the neurons or astrocytes [
69,
70]. Furthermore, cGAS is considered to be an IFN-stimulated gene (ISG); hence, it is likely that any cell with even low basal expression could upregulate this pathway in the disease contexts [
97]. For instance, in a TBI model, the expression of STING and other ISGs were upregulated 2 months post-injury, while cGAS remained unaltered in comparison to sham levels [
98]. Hence, evaluating the expression of cGAS and STING at baseline and during disease states over a period of time by using advanced techniques such as single-cell sequencing might be beneficial. Furthermore, current studies performed to determine the expression patterns of cGAS and STING are limited to rodents. Studies utilizing gyrencephalic animals (ferrets and non-human primates) and human samples need to be performed to validate the findings and study the expression patterns between species.
Second, considering the natural history of the disease (AD), the time course of cGAS and STING activation varies at different time points, and the subsequent window for treatment should be thoroughly characterized. In the context of AD, the main aim is to prevent inflammation and neuronal death. However, it is likely that there are critical times when cGAS–STING activation might be beneficial. Hence, a better understanding of the time of activation of cGAS or STING during the disease process will aid in providing therapeutic benefits.
Third, variability between species can hamper drug discovery and development because an unexpected species-specific role for STING as a receptor needs consideration. For instance, considerable molecular differences are noted between murine STING and human STING, and some agonists for murine STING are ineffective in activating human STING [
99]. A study by Conlon et al. showed that DMXAA, a murine STING agonist, showed excellent antitumor activity in mouse models but in clinical trials it failed to bind to human STING because it acts specifically against murine STING. However, rat STING appears to display similar signaling profiles towards STING agonists when compared to human STING, suggesting that the rat is more suitable for the preclinical testing of STING-targeted therapies [
100]. Using multiple animal models, including those having a gyrencephalic brain such as ferrets or non-human primates, may be beneficial for assessing cGAS–STING as a therapeutic target due to its species variability. Since none of the STING inhibitors has entered AD clinical trials, careful evaluation of blood–brain barrier permeability and side effects (infection and cancer induction) is needed [
101].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24098151