The apelin/ELABELA-APJ axis can represent a potential HF therapeutic target and has been shown to play a role in the pathophysiology of both MI and hypertension. However, one of the challenges in using the native apelin and ELABELA (ELA) peptides as therapeutic agents is their short half-life in the blood circulation or extracellular environment.
2. APJ and Its Endogenous Agonists
2.1. APJ
Apelinergic system history began in 1993 with the first description of the apelin receptor (APJ, also known as APLNR) based on its 31% homology with angiotensin II type 1 receptor (AT1R) [
3]. Since the homologous sequences are primarily present in the seven hydrophobic transmembrane domains, APJ does not bind to angiotensin II (AngII) and for this reason was considered an orphan receptor for many years. At the same time, tissue distribution of APJ and AT1R is quite similar. APJ is widely expressed in the body and highly expressed mainly in the cardiovascular (CV) system where this receptor is present in cardiomyocytes (CMs), endothelial cells (ECs), and vascular smooth muscle cells (SMCs) [
4,
5].
The amino acid sequence of this receptor is made of 377 amino acid residues and is well conserved; in fact, the human APJ gene (APLNR) located on chromosome 11q12 displayed more than 90% sequence homology to murine APJ [
6]. N-terminal glycosylation of APJ is essential for the proper receptor’s stability, protein folding, and binding to the ligand, while C-terminal palmitoylation favours the association to the cell membrane. Moreover, palmitoylation combined with phosphorylation allows APJ internalisation, dimerization, and the interaction with ligands [
7]. APJ is a G-protein-coupled receptor (GPCR), and G proteins are formed by three different subunits (α, β, and γ) and are classified according to the α subunit in four families (i.e., Gαi/o, Gαq/11, Gαs, and Gα12/13) that trigger several different signalling pathways [
8]. Upon C-terminal phosphorylation of APJ by GPCR kinases, β-arrestin is recruited and inhibits APJ activation by promoting its internalization [
9]. It is also interesting to note that in the heart APJ may act as a mechanosensor for stretch through recruitment of β-arrestin [
10].
The role of APJ resulted as pivotal in the cardiovascular system. During embryogenesis, APJ is expressed in mesoderm and APJ deficiency-caused defects in coronary vessel development [
11,
12]. In adults, APJ activation led to vasodilatation resulting in lowered blood pressure, angiogenesis, haemostasis, and anti-thrombotic effects. In the heart, it increases conduction velocity within CMs, has antiarrhythmic properties, and reduces myocardial hypertrophy and fibrosis [
8].
APJ expression increases after ischaemia through the hypoxia-inducible factor-1 (HIF-1a) pathway [
13].
2.2. Apelin
Apelin was the first discovered natural endogenous ligand of APJ. Apelin was initially isolated from a cow’s stomach in 1998 by Tatemoto and co-workers [
14], and afterwards, the presence of apelin mRNA has been detected in numerous tissues and organs, including adipose tissue, brain, liver, kidneys, skeletal muscles, heart, vessels, and lungs [
15,
16,
17]. Apelin is encoded by a gene located on chromosome Xq 25–26 in various animal species including humans, mice, rats, and cows [
16]. Apelin is initially produced as a preproprotein containing 77 amino acids, with the active sequence located in the C-terminal region. The removal of 22-unit signal peptide from N-terminal preproprotein generates the proprotein, also known as apelin-55 [
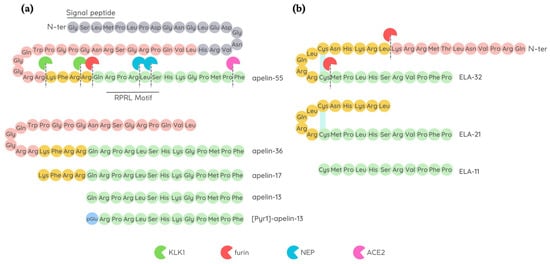
18]. Endopeptidases further cleave apelin-55 into bioactive isoforms called apelin-36, -17, or -13, accordingly to the number of amino acid residues (
Figure 1). Among them, the shorter peptides apelin-17, -13, and the pyroglutamate modified form of apelin-13 called [Pyr1]-apelin-13 have a higher affinity for APJ [
14], while fragments shorter than 10 amino acids are biologically inactive [
19]. Difference in isoform distribution was also observed; for example, [Pyr1]-apelin-13 is predominant isoform in the heart and plasma due to its resistance to degradation by peptidases [
20,
21], while apelin-36 is mainly detected in the lung, testis, and utero [
22].
Figure 1. Amino acid sequence of apelin and ELABELA (ELA) peptides. (a) Schematic representation of apelin-13, apelin-17, apelin-36, and [Pyr1]-apelin-13 that may all be produced by further processing of apelin-55; all the isoforms have the ability to bind APJ. Angiotensin-converting enzyme type II (ACE2), furin (also known as PCSK3), kallikrein (KLK1), and neprilysin (NEP) degrade native apelin isoforms, thus affecting their bioavailability; (b) Schematic representation of ELA peptide maturation by furin convertase to generate ELA-21 and ELA-11 fragments. Disulfide bridges are shown as light blue lines.
Apelin isoforms display a brief half-life in vivo, lasting less than 5 min [
23,
24] due to their rapid breakdown by different proteases. Angiotensin converting enzyme II (ACE2) is a carboxypeptidase that was firstly identified as apelin-degrading enzyme. ACE2 cleaves the Pro12–Phe13 peptide bond at C-terminal level, generating inactive apelin isoforms [
22,
25,
26]. Later, other two cleavage sites within the apelin sequence were identified in apelin-13: Arg4–Leu5 and Leu5–Ser6 at C-terminus (RPRL motif), which are cleavage sites of a zinc-dependent metalloprotease called neprilysin (NEP) [
27], thus abolishing apelin binding to APJ [
28]. Recently, prolyl carboxypeptidase (PRCP), another protease capable to hydrolyse [Pyr1]-apelin-13, has been identified in endothelial cell membrane [
29] while the action of plasma serine protease kallikrein B1 (KLKB1) on the first three N-terminal residues of apelin-17 result in the production of an inactive C-terminal 14-mer [
30].
Apelin/APJ axis plays a pivotal role in maintaining CV homeostasis [
8,
31].
In healthy humans, plasma apelin account for 0.26 ± 0.03 nmol/L concentration, and this low level would suggest a paracrine and autocrine activity [
32]. However, hypoxia conditions may raise the plasma level of apelin, and this synthesis is mediated by HIF-1 binding to the apelin gene [
33,
34,
35].
2.3. ELABELA
The discrepancy found in the foetal phenotypes between apelin knock-out and APJ-deficient mice, with observation of improper cardiac vessel formation and prenatal mortality, respectively, later led to the identification of Apela/ELABELA/Toddler (hereafter referred to as ELA) as a second APJ ligand [
36,
37].
It is encoded by the Apela/ELABELA/Toddler gene on chromosome 4q32.3, which is activated since the early embryonic stage. Apela gene encodes 54 amino acids pre-proprotein from which the active peptides ELA-32, -22, -21, and -11 (referring to the number of remaining amino acid residues at the C-terminus) are generated [
36,
38] (
Figure 1). The C-terminal 13 residues of ELA are highly conserved across species and are required for APJ binding and the consequent activation of Gi protein alpha subunit (Gαi) as well as β-arrestin signalling pathways [
38,
39].
ELA-32 and ELA-11 differ in their membrane-interactive properties, providing a potential mechanism for distinctive signalling outcomes [
40].
ELA is highly expressed in embryos, but is also present in some adult tissues, i.e., heart and blood vessels [
32,
41,
42]. Like apelin, ELA displays a short half-life in plasma, and furins have been proposed as possible proteases able to cleave ELA-55 in two conserved di-arginine motifs [
37,
38].
The absence of the ELA gene causes early abnormalities in heart development, as observed in APJ-knockout animals [
37], thus highlighting a crucial role of ELA in cardiac development and also leading to suggest a potential role of ELA in cardiac regeneration. In the rodent heart, ELA is predominantly expressed in the non-CM fraction, suggesting that ELA expression mainly occurs in fibroblasts and endothelial cells [
42]. Paracrine and autocrine activities were suggested because of its low plasma level in humans (0.34 ± 0.03 nmol/L) [
32], while in mouse heart, samples obtained 4 weeks after MI ELA expression resulted enhanced in the left ventricle of about 6 folds compared to control [
42].
Due to the rather recent discovery of ELA, there is still much to find out about its endogenous isoform functions and their interaction with APJ.
2.4. Physiological Ligand Activities in CV System
The apelin/ELA-APJ pathway is already involved in the embryonic heart development, even though the two endogenous ligands take part to this process at different time points or areas of the foetal heart as already reviewed by Kuba and co-authors [
43]. In the adult CV system, upon binding to the APJ receptor, apelin and ELA exert mostly similar biological effects.
After APJ binding, both apelin and ELA, at nanomolar concentration, exert positive inotropic effect on the in vivo rat hearts with comparable enhancement in the maximal rate of rise of left ventricular pressure (dP/dtmax), fractional shortening (c), heart rate, and cardiac output. Moreover, they reduce both left ventricular end diastolic pressure (LVEDP) and left ventricular end systolic pressure (LVESP), the latter likely due to a reduction of afterload (i.e., the resistance to cardiac output) caused by vasodilation [
32,
38,
44]. Apelin expression appeared to be crucial for proper heart function as proved by the studies of Kuba and colleagues where the lack of apelin gene caused systolic dysfunction and the gradual decline in cardiac contractility, despite the absence of histological abnormalities [
45]. Apelin-induced increase in contractility was extensively studied and showed the involvement of several pathways, among which the activation of pro-survival kinases PKC and ERK1/2 that favour the opening of sodium (NHE-1) and Ca
2+ (NCX) channels [
46,
47]. Regarding ELA-dependent inotropic effects, ELA-32 was studied by Perjés and colleagues who attributed the increase in contractility to MEK1/2-ERK1/2 but not to PKC, whereas the apelin inotropic effect was mediated by both ERK1/2 and PKC [
42]. However, the ELA-mediated inotropic effect was not completely suppressed after the inhibition of the MEK1/2–ERK1/2 pathway [
42], thus suggesting that other additional pathways can be activated by this peptide. Further investigation will be needed to clarify this issue. Remarkably, in the healthy hearts, apelin treatment exerted a moderate inotropic effect that only lasted few minutes, while the inotropic effect resulted as particularly marked when administered in failing hearts [
48,
49].
More recently, a significant increase in lusitropic effect in response to apelin treatment was observed in mouse apelin knock-out CMs [
50]. Apelin-induced positive lusitropic effect can be attributed to the preservation of SERCA activity avoiding the enhancement of intracellular calcium level, which may cause calcium overload-induced arrhythmogenesis [
50,
51].
Apelin and ELA are also vasoactive peptides with different mechanisms of action. Their administration results in a reduction in vascular resistance with consequent increase in blood flow accompanied by arterial pressure decrease. Apelin usually induces endothelial-induced vasodilation mediated by NO release via Akt activation [
23,
52,
53], while in the case of endothelial disfunction, the peptide acts directly on smooth muscle cells triggering the opposite effect [
54,
55]. On the contrary, ELA-induced vasorelaxation did not require NO release [
37]. Recently, the mechanisms of ELA-induced vasodilation have been elucidated by Sahinturk and collaborators who reported that endothelial-dependent vasorelaxation is mediated by prostanoids, while AMPK, PKC, and calcium-activated potassium channel activation was observed in the endothelial-independent pathway [
56]. It is likely that beside the apelin/ELA-mediated increase in contractility, these peptides improved cardiac function by inducing vasodilation. In fact, the lowered afterload and the increased venous return (preload) allowed the heart to enhance the stroke volume. These APJ ligand effects can be useful in hearts with progressive function deterioration.
Notably, both peptides are involved in the angiogenic process (see below). Moreover, their ability to attract and promote migration of endothelial cells led to consider apelin and ELA as chemotactic peptides in the angiogenic process [
57,
58].
APJ knockout mice exhibit abnormal body fluid balance [
59] demonstrating the involvement of apelin and ELA in water homeostasis. Indeed, they reduce water intake, and apelin also increases water excretion [
60,
61]. Moreover, apelin also resulted as involved in energy metabolism by increasing glucose uptake by tissue cells and insulin sensitivity and mitochondrial bioactivity, as recently reviewed by Hu and collaborators [
60].
3. Cardioprotective Role of APJ Endogenous Ligands
In recent years, a growing body of evidence has highlighted the potential of apelin and ELA as therapeutic targets for the treatment of CV diseases, such as hypertension, ischaemic heart disease, and heart failure.
3.1. Protection against Hypertension
Hypertension is a chronic medical condition in which the long-term high arterial blood pressure represents a relevant variable risk factor for cardiovascular disease morbidity and mortality because of increased incidence of MI and heart failure development [
62,
63]. Indeed, the persistence of high blood pressure induces LV hypertrophy and remodelling as adaptive response to cardiac output resistance [
64].
The renin–angiotensin system (RAS) is a key pathway in the development and progression of hypertension. AngII, upon binding to AT1R, exerts a vasoconstrictor effect that plays a physiological role in arterial blood pressure control through the RAS. However, AngII–AT1R axis hyperactivation commonly leads to development and progression of hypertension [
65]. AngII is formed starting from liver-produced angiotensinogen precursor which is cleaved by renin into Angiotensin I which in turn is converted to AngII through the catalytic activity of the angiotensin converting enzyme (ACE). Conversely, ACE2 negatively regulate RAS through the conversion of AngII into Ang(1–7), resulting in blood pressure reduction [
66].
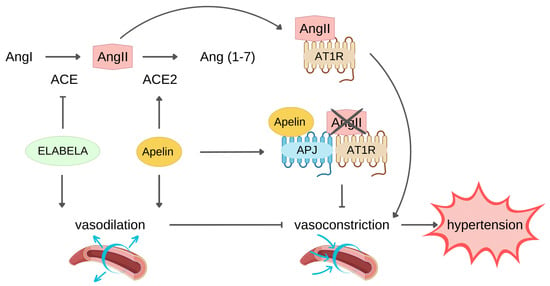
The apelin/ELA-APJ system can affect hypertension counteracting RAS (Figure 2).
Figure 2. ELABELA and apelin protect against hypertension counteracting the renin-angiotensin II system (RAS) with different mechanisms. Angiotensin Converting Enzyme (ACE) converts Angiotensin I (AngI) to Angiotensin II (AngII), which binds Angiotensin II Receptor Type 1 (AT1R) and induces vasoconstriction, leading to hypertension. Endogenous ELABELA and apelin neutralize AngII-induced vasoconstriction through their direct vasodilator effect. Both ligands exert vasodilation through AngII level reduction: ELABELA downregulates ACE expression, limiting AngII production, while apelin boosts the expression of ACE2 which hydrolyzes AngII to Ang(1–7). The binding of apelin to its receptor APJ leads to the heterodimerization of APJ and AT1R, decreasing the AT1R availability for binding AngII, thus inhibiting the AngII-AT1R signalling pathway that leads to hypertension.
Indeed, apelin/ELA and AngII exerted an opposite effect on vessels. In this regard, it has been reported that administration of either apelin or ELA had a blood pressure-lowering effect, potentially indicating a therapeutic role of these peptides in reverting hypertension, thus reducing the risk of developing HF [
67,
68]. In addition, apelin is capable of boosting ACE2 expression, and this is another mechanism by which it counteracts RAS. In fact, Sato and colleagues pointed out that apelin improved ACE2 promoter activity in neonatal CMs in a dose-dependent manner [
69]. In line with the in vitro results, apelin KO mice also showed downregulation of ACE2 expression and, consequently, low ACE2 protein level [
69,
70]. Of note, ACE2, which primarily acts on AngiII, has a role in apelin metabolism because it can cleave apelin into inactive isoforms, though it is unable to degrade ELA [
23,
66,
67]. Conversely, ELA did not affect ACE2 expression, but it reduced angiotensin-induced blood pressure via downregulation of ACE expression.
Another apelin mechanism opposing RAS consists of heterodimer formation by APJ and AT1R interaction, resulting in inhibition of AngII binding to its receptor and signalling pathway activation [
71].
Protracted hypertension induces remodelling of the heart which consists of cardiac hypertrophy and deposition of collagen fibres between the CMs, namely interstitial fibrosis. This morphological alteration also contributes to cardiac dysfunction, and indeed, CMs develop hypertrophy as adaptation to elevated cardiac workload, with the aim of maintaining left ventricle ejection fraction (LVEF) unchanged, at least at the beginning of the pathological condition. This protracted cardiac remodelling can give rise to arrhythmogenesis and HF over-time [
72].
The link between the apelinergic pathway and AngII RAS pathways was studied in apelin−/−mice through subcutaneous infusion with AngII for two weeks. AngII treatment caused increased cardiac dysfunction, hypertrophy, and fibrosis and enhanced ACE/ACE2 as well as TGF-β expression in apelin−/−compared to WT mice [
73]. It was also observed that the loss of apelin facilitated AngII-induced injury pathways, leading to enhanced pathological hypertrophy and myocardial fibrosis [
70,
74]. Furthermore, apelin-deficient mice showed downregulation of ACE2, increased superoxide production, and augmented NADPH oxidase activity [
70].
In another animal model, AngII-induced hypertension yielded an increase in myocardial fibrosis and inflammation. Moreover, AngII lowered apelin and ELA levels in both rat and primary neonatal rat cardiac fibroblasts, and these results were in line with the low level of these peptides in hypertension patients [
75]. The adverse remodelling was shown to be mediated by miR-122p, as the treatment of primary neonatal rat cardiac fibroblasts with both AngII and the inhibitor of miR-122p abolished the AngII-induced downregulation of APJ ligands [
75]. Interestingly, pre-treatment of cardiac fibroblasts with apelin or ELA prevented the enhancement of inflammation and fibrosis markers. These authors also highlighted a link between APJ ligand level and cardiac fibrosis [
75].
On the other hand, the animal treatment with apelin-13 could counteract AngII-induced fibrosis via TGF-β1/SMAd2/α-SMA and preserved the homogeneity of electrical conduction [
76].
In in vivo studies, the AngII-dependent ELA reduction increased cardiac hypertrophy, fibrosis, and dysfunction in murine heart, while ELA administration reduced cardiac fibrosis, ultrastructural injury, lipid peroxidation, and inflammation, thus improving cardiac function [
77]. Moreover, ELA mice administration for two weeks after transaortic constriction (TAC) promoted ACE expression downregulation and reduced cardiac fibrosis, promoted the maintenance of FS rate, and suppressed the expression of cardiac hypertrophy markers, such as BNP, ANF, and β-Myhc [
78].
Notably, ELA and apelin circulating levels were found decreased in patients with hypertension [
79,
80,
81]. Additionally, Baysal and colleagues discovered that hypertensive individuals with impairment in left ventricular systolic and diastolic performance displayed low plasma apelin level that increased following one month of antihypertensive medication [
82]. Further, concerning the ELA level, it was observed that reduced ELA plasma concentration was associated with atrial fibrillation in hypertensive patients [
83]. Among the HF patients, those with higher level showed a more preserved LVEF and better outcomes [
84]. Furthermore, low apelin and ELA expression was found in patients with pulmonary arterial hypertension [
32].
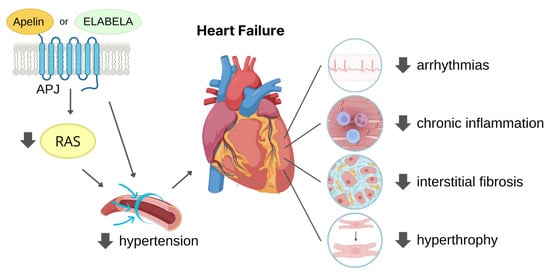
Taken together, these findings suggest that APJ ligands have significant potential as therapeutic strategy to counteract hypertension and the related complications (Figure 3).
Figure 3. Dual mechanism of ligand-activated APJ in reducing hypertension-induced heart failure. The figure illustrates the two ways in which APJ activation can reduce cardiac overload-induced heart failure. Firstly, by reducing renin-angiotensin system (RAS) activation, APJ lowers blood pressure, thus decreasing cardiac afterload. Secondly, activated APJ directly acts on the heart, decreasing the risk of developing arrhythmias chronic inflammation, interstitial fibrosis, and cardiac hypertrophy, leading to improved cardiac function.
3.2. Protection against MI
MI is a critical cardiovascular event associated with high morbidity and mortality in which blood flow blockade prevents the O
2 supply to the cardiac tissue. The restoration of blood flow, via thrombolytic therapy, primary percutaneous coronary intervention, and coronary artery bypass grafting (CABG), is essential for cardiac tissue salvage. However, after reperfusion, an exacerbation of myocardial damage takes place. For this reason, it is more correct talking of ischaemia-reperfusion (I/R) injury that consists of a series of events including cell death, inflammation, oxidative stress, calcium overload, and paradoxical pH that lead to cardiac tissue damage as well as impaired heart function (i.e., stunning of myocardium) [
85]. Notably, it has been estimated that about 30% of patients develop I/R injury which seriously affected patient prognosis [
85,
86].
4. APJ Peptide Agonists
Modified peptides can be designed to specifically bind a target receptor. Peptide modifications might modulate protein–protein interactions to improve target selectivity and affinity that play crucial roles during the persistent interaction between proteins. Furthermore, compared to small molecules and antibodies, modified peptides have the important advantage of causing a minor immune response [
166]. The small size of APJ peptide ligands makes them easy to manipulate.
4.1. APJ Endogenous Ligand Main Chain Modifications
The well-known short in vivo half-life that characterises both apelin and ELA poses a hurdle to their clinical translation. Many studies thus aim to develop more stable APJ ligands, based on either apelin or ELA peptide structure, through many different approaches, such as main-chain residue removal/addition, palmitoylation, and cleavage site modification of the several proteases involved in the degradation of parental peptides to obtain longer half-life in blood circulation as well as in tissues, trying to maintain at the same time apelin- or ELA-like cardiovascular effects (Table 1 and Table 2).
Table 1. Main chain modifications of APJ ligands, based on [Pyr1]-apelin-13, apelin-12, apelin-17, or ELA peptide, and their effects on stability, binding affinity, and cardiovascular properties.
Table 2. Peptides obtained by apelin cyclization and their effects on plasma stability, binding affinity, and cardiovascular properties.