Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Cancer stem cells (CSCs) are a small subpopulation of cells within tumors with properties, such as self-renewal, differentiation, and tumorigenicity. CSCs have been proposed as a plausible therapeutic target as they are responsible for tumor recurrence, metastasis, and conventional therapy resistance. Selectively targeting CSCs is a promising strategy to eliminate the propagation of tumor cells and impair overall tumor development.
- cancer
- cancer stem cells
- colon cancer
1. Introduction
Cancer malignancy relates to tumor heterogeneity, and it has been suggested that it is driven by a minor subpopulation of cells called cancer stem cells (CSC) [1][2]. CSCs are a small subpopulation of cancer cells that can self-renew, differentiate, and start tumor growth, thereby mediating drug resistance and cancer progression, resulting in supporting tumor recurrence and distant metastasis [3][4][5]. Furthermore, CSCs can differentiate into different cell populations with a high plasticity potential, they have a high resistance to stressful conditions in the tumor microenvironment (TME), they can induce cell death by chemotherapeutic agents [6], and have the ability to invade healthy tissues. All of these characteristics are related to tumor progression, metastasis, and resistance to antitumoral therapies [1][7].
Surgery, radiotherapy, and chemotherapy are conventional treatments. Surgery can successfully remove cancer masses from the body, while combining radiotherapy with chemotherapy can effectively lead to better results for treating numerous types of cancer [7]. Nevertheless, even when conventional cancer treatments target the bulk of the tumor, they cannot eliminate CSCs responsible for metastasis and tumor recurrence. Therapeutic alternatives to chemotherapy and surgery, such as cell therapy against CSCs, have yielded results in recent years. CSCs’ surface markers are well characterized, making possible the creation of bio-targeted therapies.
Cell therapy is a promising option in bio-directed cell-targeted therapies against CSCs, particularly with modified cells that enhance the interactions and mechanisms related to the eradication of CSCs. These cellular therapies against CSCs work in different ways: (i) they can release factors that modulate and regulate the TME and enhance the suppression of cancer cells [8] through the activation or inhibition of some signaling pathways; (ii) they can act as a cell-targeted delivery system for anticancer drugs [9], and (iii) they can induce cell apoptosis through the receptor-binging union.
2. Specific Targets Used in CRC Cell Therapy with Potential Antitumoral Effect against CSCs
There are specific targets in CSCs that can be used as therapeutic targets, such as CD44, CD133, CD24, and ALDH; however, these markers are not frequently used in cell therapy because they are commonly expressed in normal stem cells. Among other disadvantages, they only represent a low percentage within the tumor and have variable expression during the disease stages and after chemotherapy or radiotherapy. CRC targets have been proposed as an attractive cell therapy strategy and stand out due to overexpression in tumor tissues compared to healthy tissue, and recently to target pathways or the activity of CSCs (Figure 1). Some of the most prominent are:
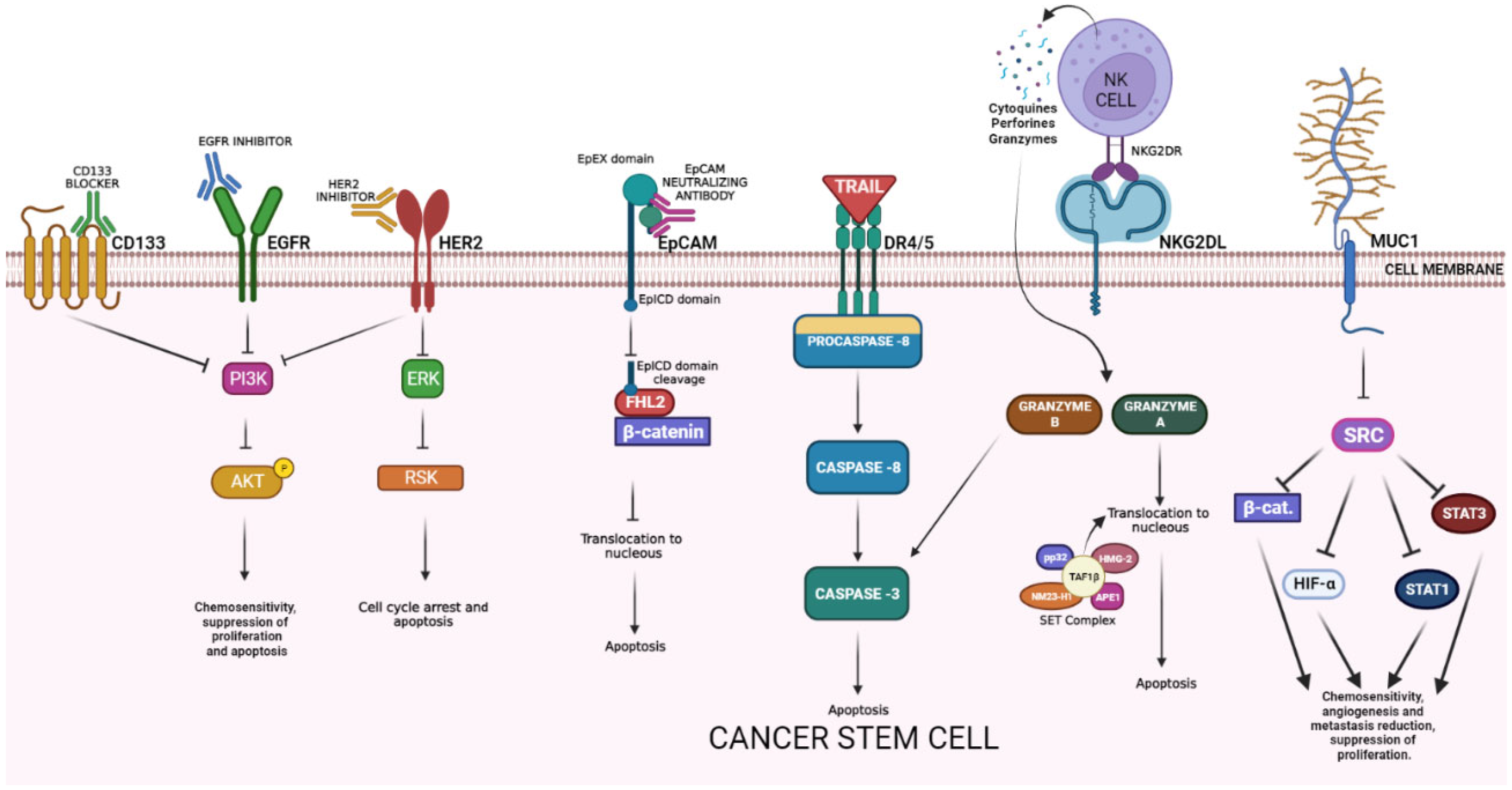
Figure 1. Cancer stem cell surface markers and their antitumoral mechanisms. EGFR = epidermic growth factor receptor; PI3K = Phosphatidylinositol 3 kinase; AKT = protein kinase B; HER2 = human epidermal growth factor receptor 2; ERK = extracellular signal-regulated kinases; RSK = ribosomal S6 kinase; EpCAM = epithelial cell adhesion molecule; EpEX = EpCAM extracellular domain; EpICD = EpCAM intracellular domain; FHL2 = four and a half LIM domains protein 2; DR4/5 = death receptor 4/5; TAF1 BETA = TATA-box binding protein associated factor 1; NKG2R = natural killer group 2 receptor; NKG2L = natural killer group 2 ligand; MUC-1 = mucin 1; HIF-α = hypoxia-inducible factor; STAT1 = signal transducer and activator of transcription 1.
EGFR is a transmembrane glycoprotein belonging to the protein kinase superfamily activated by ligand binding, that causes receptor dimerization and activates multiple pathways that leads to gene activation for cell survival, proliferation, and differentiation. It is also related to functions of metabolism and affects autophagy. Some reports suggest that it can sensitize CSCs to apoptosis by chemotherapy (5FU) after treatment with EGFR monoclonal antibody. [10] An in vivo model showed that anti-EGFR therapy can affect CSC numbers and counteract the chemotherapy-induced CSCs’ expansion [11]. EGFR inhibitors suppressed proliferation and induced apoptosis of CSCs by inhibiting autophosphorylation of EGFR and downstream signaling proteins, such as Akt kinase and extracellular signal-regulated kinase 1/2 (ERK 1/2) (Figure 1) [12].
HER2 is a non/ligand-binding member of the EGFR family. It homodimerizes or heterodimerizes with other EGFR family members (HER1/EGFR, HER3, HER4), inducing the transphosphorylation of the intracytoplasmic tyrosine kinase domain and activation of a variety of downstream signal transduction pathways (e.g., RAS/RAF/ERK, PIK3K/AKT/mTOR, JAK/STAT3). The overexpression of HER2 causes hyperactivation of mitogenic signals, resulting in uncontrolled cell proliferation and cancer. HER2 amplification and protein overexpression can be identified in approximately 6% of CRC patients [13][14][15][16][17]; both HER-2 and HER-3 are overexpressed in liver metastasis in CRC patients (8% and 75%, respectively) [18] HER2 amplification most commonly occurs in the rectum and has been linked to the resistance of EGFR-targeted therapy and lower overall survival compared to HER2 wild-type CRC [19][20]. Early onset research in the area determined that the expression of the markers CD24, CD44, ALDH, and regulated epithelial-mesenchymal transition (EMT) is seen in the presence of HER2 blockage in breast cancer [21]. HER2-overexpressing gastric cancer cells exhibited increased stemness and invasiveness and were regulated by Wnt/β-catenin signaling [22].
The epithelial cell adhesion molecule (EpCAM) is a type I transmembrane glycoprotein expressed mostly in the basolateral membrane of normal epithelial cells [23]. Under normal conditions, EpCAM is involved in cell-cell adhesion and the regulation of differentiation in progenitor and embryonic stem cells; however, in the context of cancer, EpCAM overexpression is related to increased cell proliferation, migration, invasion, and tumor metastasis [23][24]. Although it has been shown to be overexpressed in a wide variety of epithelial tumors, EpCAM seems to be associated with a poor prognosis in certain cancer types (colorectal, breast, prostate, gallbladder, ovarian, bladder, pancreas, and adenoid cystic carcinomas). EpCAM has been reported as a marker of better prognosis in other tumors (esophageal, renal, gastric, endometrial, thyroid, and head-and-neck carcinomas) [25]. In CRC, EpCAM is overexpressed in over 90% of all cancer cells [24]. The EpCAM neutralizing antibody exhibited antitumor effects via inhibiting the nuclear translocation of EpICD/β-catenin complexes and induced apoptosis in colon cancer cells (Figure 1) [26]. EpCAM is expressed in both healthy and cancer tissues; however, it is usually overexpressed in cancerous tumors derived from epithelial tissues [26]. Based on this fact, immunotherapies have been developed using monoclonal antibodies against EpCAM. To increase the efficacy and specificity of EpCAM treatment, combinatorial approaches have been developed, where the EpCAM antibody is fused with another anticancer molecule that targets a different cancer receptor/ligand, and improves the specificity for cancer cells, such as catumaxomab (Removab), which combines anti-CD33 antibody and anti-EpCAM antibody, and increases the specificity of the therapy [26][27]. Lastly, most therapies are locally infused to reduce the damage to healthy tissue.
Mucin 1 (MUC-1) is a heterodimeric transmembrane glycoprotein. Its function is related primarily with the formation of a physical barrier to lubricate and protect normal epithelial tissues and mediate signal transduction [26]. It is upregulated in response to inflammation, and is aberrantly overexpressed in diverse types of cancer [27][28]. The MUC1 molecule consists of an N-terminal subunit (MUC1-N) and C-terminal transmembrane subunit (MUC1-C) [29]. MUC1 can act as a cell surface antigen to colorectal CSC [30] resistance and angiogenesis of tumor development. Recent studies have supported a previously unreported function for MUC1-C in activating PRC2 and PRC1 in cancer cells. In the regulation of PRC2, MUC1-C drives the transcription of the EZH2 gene, binds directly to EZH2, and enhances the occupancy of EZH2 on target gene promoters with an increase in H3K27 trimethylation. Regarding PRC1, which is recruited to PRC2 sites in the hierarchical model, MUC1-C induces BMI1 transcription, forms a complex with BMI1, and promotes H2A ubiquitylation. MUC1-C thereby contributes to the integration of the PRC2 and PRC1-mediated repression of tumor suppressor genes, such as CDH1, CDKN2A, PTEN, and BRCA1. Just as PRC2 and PRC1, MUC1-C is associated with the EMT program, CSC state, and acquisition of anticancer drug resistance. In concert with these observations, targeting MUC1-C downregulates EZH2 and BMI1, inhibits EMT and the CSC state, and reverses drug resistance. These findings emphasize the significance of MUC1-C as a therapeutic target for inhibiting the aberrant PRC function and reprogramming the epigenome in human cancers [29][31].
The natural killer group 2, member D (NKG2D) is a stimulatory receptor found in some cytotoxic immune cells that recognizes the NKG2D ligands (NKG2DL) [7]. These molecules are not expressed in normal cells, but are upregulated in stressed and many malignant cells. In humans, two distinct categories of NKG2DL are expressed, one family consists of the major histocompatibility complex (MHC) class I polypeptide-related sequence A and B (MICA and MICB, respectively), while the other family of NKG2D ligands consists of a six-member glycoprotein family of UL16-binding proteins (ULBP1–6) [8]. In normal conditions, CSCs exhibit a lower expression of MHC-I type NKG2DL and a higher expression of UL16-binding proteins NKG2DL [9], but when there is DNA damage or cell cycle alterations, some MHC-I type NKG2DL becomes upregulated and expressed into CSCs [8]. NKG2D ligands are recognized by NKG2D receptors on subsets of neighboring cytotoxic immune cells (natural killer (NK) cells, natural killer T (NKT) cells, subsets of gamma delta (γδT cells). The natural killer group 2D (NKG2D) has recently emerged as a major activating receptor on T lymphocytes and natural killer cells. In both humans and mice, multiple genes encode ligands for NKG2D, and these ligands are non-classical MHC Class I. The NKG2D-ligand interaction triggers and activates a signal in the cell expressing NKG2D, and this promotes cytotoxic lysis of the cell expressing the ligand. Most normal tissues do not express ligands for NKG2D, but ligand expression has been documented in tumor and virus-infected cells, leading to the lysis of these cells. The tight regulation of ligand expression is important. If there is inappropriate expression in normal tissues, this will favor autoimmune processes, whilst failure to upregulate the ligands in pathological conditions would improve cancer development or the dissemination of intracellular infection [32].
TRAIL is a type II transmembrane protein denominated tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL, CD253). It has been widely studied as a strategy for tumor elimination since cancer cells overexpress TRAIL death receptors, selectively inducing apoptosis [33]. In contrast with most chemotherapeutic drugs, this protein triggers the extrinsic apoptotic pathway in malignant cells in a p53-independent manner. TRAIL binds to death receptors DR4 (TRAIL-R1) and DR5 (TRAIL-R2), decoy receptors DcR1 and DcR2, and osteoprotegerin (OPG). The binding to its DR4 and DR5 receptors activates the homotypic DD-dependent recruitment of Fas-associated protein with death domain (FADD). This protein bridges pro-caspases 8 and 10 to form a death-inducing signaling complex (DISC), that activates caspases-8, 3, and 7, leading to apoptosis. In vitro, TRAIL can induce apoptosis in a wide variety of tumor cells. One of the main limitations of the recombinant protein TRAIL application is fast the clearance from the serum [34]. Moreover, it was observed that soluble TRAIL and monoclonal antibodies against DR4 and DR5 present potent antitumor activities in in vivo models without systemic toxicity [33]. Recombinant TRAIL treatment has been evaluated in clinical trials. Although it has had an important antitumor effect, it has a short half-life and repeated applications are needed to obtain the desired effect or systems that facilitate its continuous expression [35].
CD133, the cluster of differentiation 133 or prominin-1, is a five-transmembrane glycoprotein expressed in several progenitor and stem cells. It participates in the organization of the topology of the plasma membrane [36]. Researchers have proposed CD133 as a cell surface marker of CSCs since its expression seems to relate to chemoresistance, an elevated risk of distant metastasis, and relapse [36][37]. Moreover, it participates in primordial cell differentiation and EMT [38][39][40]. CSCs display an EMT phenotype, possess elevated levels of the transcription factors SNAIL and TWIST, the mesenchymal markers vimentin and fibronectin, and low levels of epithelial protein E-cadherin [41]. CD133+ CRC cells manifest CSC-like properties, such as higher levels of the SC markers OCT4 and SOX2, a tumor sphere-forming ability, and are more tumorigenic in NOD/SCID mice [41]. This finding is consistent with OCT4 and SOX2 overexpression in poorly differentiated human tumors [42]. CD133+ CSCs might escape immune surveillance by expressing the inhibitory molecule B7H1. Moreover, the innate immune system can effectively be recruited to kill CSCs using bispecific antibodies targeting CD133 [43]. CD133 can upregulate the expression of the FLICE-like inhibitory protein (FLIP) in CD133-positive cells, inhibiting apoptosis. In addition, CD133 can increase angiogenesis by activating the Wnt signaling pathway and increasing the expression of vascular endothelial growth factor-A (VEGF-A) and interleukin-8. Therefore, CD133 could be an “Achilles’ heel” for CSCs, because by inhibiting this protein, the signaling pathways that are involved in cell proliferation will also be inhibited (Figure 1) [44].
3. Cell Types Used in CRC Immunotherapy
3.1. T-Cells
T cells are part of the adaptive immune system, divided into CD4+ (helper) and CD8+ (cytotoxic) T cells. CD4+ T cells support the body´s adaptive response to different classes of pathogens by cytokine production. CD8+ cytotoxic T lymphocytes are activated in response to tumor-associated antigens present in the context of MHC class I molecules [45]. T cell immunotherapy has different strategies, such as increasing or inhibiting cellular immunity or inducing changes in T cell receptors to recognize specific targets.
Chimeric antigen receptor (CAR) T cell receptors are designed to (1) deliver strong activation, proliferation, and survival signals via a single binding event; (2) in an independent manner to MHC, bypassing the MHC downregulation by certain tumors; and (3) exhibiting a high affinity even at low antigen density. Techniques involving CAR T cell methods have evolved from the basic design, ectodomain antibody single-chain fragment, and variable fragment engineered to the T cell receptor (TCR)-chain to multiple CAR generations. As a result, CAR design has progressed from simple molecules to more complex moieties, allowing for the combinatorial antigen selection with diverse signaling properties. This advancement facilitates the development of armored CAR T cells with improved antitumor activity and good toxicity management.
3.1.1. Checkpoint Inhibitor Drugs
T cell activity can be controlled by checkpoint inhibitor drugs that hinder the blocking of tumor-associated immunosuppression and allow cytotoxic cells and lymphocytes to attack tumor cells. However, in advanced CRC, immune checkpoint therapy is limited to patients with high microsatellite instability (MSI-H) (approximately 5%). This type of CRC is associated with high rates of tumor mutation (tumor mutational burden high—TMB-H) and tumor-infiltrating lymphocytes [45].
Tumor-infiltrating lymphocytes expressing Fas are very susceptible to Fas-mediated apoptosis. Thus, inhibition of FasL on colon cancer cells improves antitumor immunity and reduces tumor growth. However, serum levels of FasL increase in colon cancer and have a decreased or mutated expression of FasR, TRAIL-R1, and TRAIL-R2 death receptors on their cell surface, promoting survival [46].
3.1.2. Adoptive T-Cell Therapy
Adoptive T cell therapy (ATC) takes patient-derived ex vivo expanded T cells and reinfuse them into patients [45]. Unlike conventional T lymphocytes that recognize peptide antigens bound to highly polymorphic MHC molecules, Vγ9Vδ2 T cells recognize nonpeptidic antigens without antigen processing and MHC restriction. Vγ9Vδ2 T cells stay preactivated, lacking antigen exposure. An in vivo administration of compounds (aminobisphosphonates and IL-12) that activate Vγ9Vδ2 T cells or an adoptive transfer of ex vivo expanded cells are necessary to drive its antitumor activity. In vitro, Vγ9Vδ2 T cells present strong cytotoxic activity against tumor cell lines or primary cells from colon carcinoma [46]. Moreover, γδ T cells are one of the most prominent immune cells in the gut and good candidates for immunotherapeutic strategies in CRC [45].
CSCs in CRC are commonly resistant to T cell therapy. As a form of sensibilization, chemotherapeutic drugs used for CRC treatment, such as 5-fluorouracil and doxorubicin, have been used in CSCs from CRC cell lines in combination with autologous Vγ9Vδ2 T cells, increasing DR5 expression and improving cytotoxic activity at low doses [46].
Adoptive cell transfer (ACT) is a form of cell therapy, in which T cells isolated from cancer patients’ tumors are modified by engineering, they are selected, expanded ex vivo, then reinfused into the patients [4]. ACT has already been clinically successful in treating hematologic malignancies; however, current evidence suggests that T cell therapy can improve treatment against CRC, even at advanced disease stages [47]. ACT therapy of ex vivo expanded αβ T cells (anti-CD3 and IL-2 stimulation) in combination with chemotherapy XELOX (capecitabine and oxaliplatin), and bevacizumab have achieved an 80% response rate and acceptable toxicity in stage IV CRC [48].
3.1.3. T-Cell Receptor Therapy
In T cell receptor therapy (TCR), the T cell receptor is modified to target a specific antigen presented by an MHC molecule. In patients with CRC, carcinoembryonic antigen (CEA) is a frequently upregulated common target antigen [45]. As another strategy, transgenic TCR can bind with CEA+ CRC cells, enhancing tumor recognition compared to wild-type T cells [49] In a study of three patients with metastatic CRC, targeted therapy of TCR reduced CEA levels between 74–99%, and one patient had metastasis regression. However, severe transient inflammatory colitis toxicity occurred [50]. Clinical trial NCT02757391 is testing CD8+ T cell therapy in combination with pembrolizumab (PD-1 inhibitor) [45].
3.1.4. Chimeric Antigen Receptor T Cell
The chimeric antigen receptor T cell or CAR-T cell is a genetically modified T cell that can avoid the MHC and directly target a surface antigen of interest. CAR receptors comprise a target-binding extracellular region that gives antigen specificity conformed by a single-chain variable fragment (scFv) antibody, a hinge and transmembrane region, and an intracellular domain related to T cell activation via the TCR CD3ζ signaling chain [51]. CAR-T cell therapy attempts to express functional chimeric receptors that recognize tumor antigens in a non-MHC-restricted manner, allowing for the recognition of any desired target. Despite its success in hematologic diseases, its response in solid tumors, such as CRC, is less than 9%, mainly due to the lack of uniformly expressed target antigens [45][52]. In solid tumors, such as CRC, surface proteins, such as the natural killer group 2, member D (NKG2D), and CEA are proposed [52].
The first clinical trial of CAR-T cells for CRC (NCT02349724) used CEA as a target. Treatment showed that 70% of patients with progressive disease and those previously treated presented stable disease for more than 30 weeks and a decreased tumor volume without severe secondary effects. Applying anti-CEA CAR-T cells in CEA+ adenocarcinoma with liver metastasis (NCT02416466 and NCT02850536) intraarterially administered, improved the delivery of cells into the metastasis and reduced cytokine release syndrome (CRS) [51]. However, before the employment of CAR-T cell therapy, several changes in the tumor microenvironment should be considered, as in a clinical trial of CEA CAR-T cells that increased the resistance of CEA+ rectal cancer tumor immunity [53].Two current phase I trials are recruiting subjects. The NCT04107142 trial uses CAR-T cells targeting NKG2DL, and NCT03970382 is testing neoantigen-targeted TCR on locally advanced or metastatic tumors. In patients, applying different quantities of autologous and allogenic NKG2D CAR-T cells showed that 1 × 108 and 3 × 108 cells were achieved without dose-limiting toxicity [52]. In NCT03692429 against NKG2D ligands, a modified TCR was used to make them suitable for allogeneic use with a TCR inhibitory molecule (TIM) sequence. Patients remained stable for approximately 3 months after treatment without graft vs. host disease [51]. Another antigen implicated in tumor growth and regulation of EMT in CRC is doublecortin-like kinase 1 (DCLK1). DCLK1-scFv (CBT-511) CAR-T cells induced cytotoxicity and increased IFN-γ release in coculture with CRC cells [45][52].
The human epidermal growth factor receptor 2 (HER2) is a potential target for CRC treatment, since it is highly overexpressed in this type of tumor. For metastatic CRC, HER2-targeted CAR-T cells eliminated numerous HER2+ solid tumors presenting signs of prevention of CRC progression in a xenograft model [47][54]. Guanylyl cyclase c (GUCY2C), a membrane-bound receptor overexpressed in more than 95% of CRC metastases, is a potential target. In a syngeneic murine model of CRC, GUCY2C CAR-T cells provided long-term protection against lung metastases [55]. However, independently of the high CAR-T cell efficiency, a problem with this therapy is the development of CRS, frequently observed by the overactivation of T cells. CRS can present simple fatigue or develop into life-threatening outcomes with a capillary leak through a severe increase of cytokines, such as IL-1, IFN-γ, and TNF-α [52].
3.2. Natural Killer (NK) Cells
Natural killer (NK) cells are lymphocytes that differ from the B and T cells belonging to the innate immune system. They originate in the bone marrow and are found in blood and lymphatic tissues, especially the spleen. Morphologically, they are large lymphocytes with cytoplasmic granules [56], and their characteristic phenotype is TCR-, BCR-, CD3-, CD16+, and CD56+. Its main functions are cytotoxicity and cytokine secretion [57]. According to the expression of CD56, they are classified into two subsets: the CD56 low/dark, which is antitumor cytotoxic, and CD56 bright [51][58] NK cells are a subset of immune effector cells that play an important role in immune activation against aberrant cells. Unlike T cell activation, NK cell activation is mediated by the contact of NK receptors with target cells. This mechanism is independent of antigen processing and presentation. An advantage of NK cells is their inherent ability to discriminate between healthy and malignant cells. NK cells express germ-line encoded activation and inhibitory receptors that trigger activation while balancing activating and inhibitory signaling. NK cells are activated by receptors that recognize stress-induced ligands on the surface of malignant cells. Normal cells express inhibitory receptors as self-major histocompatibility complex (MHC) class I molecules [59].
Another type of NK cells is cytokine-induced cells (CIK) [53][60], a heterogeneous population of NKT cells co-expressing CD3 and CD55 derived from T cell precursors. NK cell-mediated targeting and destruction of malignant cells is defined by an interplay of signals generated by inhibitory and activating NK cell receptors, that interact simultaneously with their ligands on target cells [61]. The ability of NK cells to destroy and eliminate target cells is determined by the balance between their activating and inhibitory signals, i.e., ligands expressed on target cells. These ligands interact with the NK cell surface receptors and trigger activating or inhibitory signals; thus, antigen specificity does not control NK cells [62]. In addition, the destruction of tumor cells by NK cells is not dependent on MHC or antibodies, these cells are attracted by the stress that characterizes TME [56]. Moreover, CSCs commonly express low or no MHC class I, which makes them susceptible to NK cell targeting. In some cases, CSCs also overexpress NK cell activating markers, such as CD24, CD44, CD133, and ALDH, facilitating its elimination by stimulating NK activation markers, such as MICA/B, Fas, and death receptors [63]. Thus, it is considered the ideal target because the expression of MHC-1 decreases, resulting in NK cell activation by a self-missing recognition process.
Various regulatory and stimulatory cytokines, such as IL-2, IL-21, IL-12, IL-8, IL-15, and interferon type 1, promote NK cell activities on tumor elimination [64]. Upon activation, NK cells release cytotoxic granules containing perforin and granzymes that directly lyse tumor cells, including activated cytotoxic T cells [65]. The NK cell receptor activator NKG2D, tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) [66], and perforin-mediated pathways [67] are important for CIK cell-mediated recognition and lysis of malignant cells.
In the history of NK cells as a treatment for neoplasms, a greater number of reports have been seen in acute megaloblastic leukemia [68], in which its effect has been evaluated by allogeneic transplantation of hematopoietic progenitors from haploidentical donors [69] in adult patients using NK lymphocytes from the donor. Focusing on the innate rather than the adaptive immune system, NK cells are not specific for antigens, such as T cells. They can bypass the engagement of tumor cell PD-L1 and T cell PD-1 to mediate direct cytotoxicity against CSCs [47]. For this reason, researchers are studying NK cells to potentiate their function in vitro before infusion in ACT as ex vivo allogeneic NK cells with a mixture of cytokines termed cytokine-induced memory-like NK cells (CIML-NK), which can combine with CAR engineering [70]. Based on the goal of inducing antigen-specific T cells in patients, using DC vaccines as antigen-presenting mechanisms that target tumor-derived blood vessels to disrupt tumor angiogenesis and decrease tumor growth is another form of immunotherapy for CRC patients [71][72]. NK cells can differentiate from induced pluripotent stem cells (iPSC). This source avoids the current requirements of collection and expansion as IPSCs can grow indefinitely by self-renewal. Furthermore, IPSC-derived NK cells are homogenous and clinically produced in a scalable manner [1]. Moreover, this approach allows for multiple genetic modifications to improve NK cell cytotoxicity [2]. There are diverse methods for genetic modification of iPSC-derived NK cells, such as lentivirus, transposons, and CRISPR-Cas9 system [3]. Thus, genetically engineered iPSC-derived NK cells could represent a promising strategy for a renewable source of NK cells for immunotherapy of solid tumors, such as CRC.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24098163
References
- Sun, H.R.; Wang, S.; Yan, S.C.; Zhang, Y.; Nelson, P.J.; Jia, H.L.; Qin, L.X.; Dong, Q.Z. Therapeutic Strategies Targeting Cancer Stem Cells and Their Microenvironment. Front. Oncol. 2019, 9, 1104.
- Kuşoğlu, A.; Avcı, Ç.B. Cancer Stem Cells: A Brief Review of Current Status. Gene 2019, 681, 80–85.
- Lin, C.C.; Liao, T.T.; Yang, M.H. Immune Adaptation of Colorectal Cancer Stem Cells and Their Interaction with the Tumor Microenvironment. Front. Oncol. 2020, 10, 588542.
- Zalewski, A.; Snook, A.E.; Waldman, S.A. Stem Cells as Therapeutic Targets in Colorectal Cancer. Pers. Med. 2021, 18, 171–183.
- Du, L.; Cheng, Q.; Zheng, H.; Liu, J.; Liu, L.; Chen, Q. Targeting Stemness of Cancer Stem Cells to Fight Colorectal Cancers. Semin. Cancer Biol. 2022, 82, 150–161.
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472.
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and Their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923.
- Aravindhan, S.; Ejam, S.S.; Lafta, M.H.; Markov, A.; Yumashev, A.V.; Ahmadi, M. Mesenchymal Stem Cells and Cancer Therapy: Insights into Targeting the Tumour Vasculature. Cancer Cell Int. 2021, 21, 158.
- Veselov, V.V.; Nosyrev, A.E.; Jicsinszky, L.; Alyautdin, R.N.; Cravotto, G. Targeted Delivery Methods for Anticancer Drugs. Cancers 2022, 14, 622.
- Feng, Y.; Gao, S.; Gao, Y.; Wang, X.; Chen, Z.; Feng, Y.; Gao, S.; Gao, Y.; Wang, X.; Chen, Z. Anti-EGFR Antibody Sensitizes Colorectal Cancer Stem-like Cells to Fluorouracil-Induced Apoptosis by Affecting Autophagy. Oncotarget 2016, 7, 81402–81409.
- De Angelis, M.L.; Zeuner, A.; Policicchio, E.; Russo, G.; Bruselles, A.; Signore, M.; Vitale, S.; De Luca, G.; Pilozzi, E.; Boe, A.; et al. Cancer Stem Cell-Based Models of Colorectal Cancer Reveal Molecular Determinants of Therapy Resistance. Stem Cells Transl. Med. 2016, 5, 511–523.
- Feng, Y.; Dai, X.; Li, X.; Wang, H.; Liu, J.; Zhang, J.; Du, Y.; Xia, L. EGF Signalling Pathway Regulates Colon Cancer Stem Cell Proliferation and Apoptosis. Cell Prolif. 2012, 45, 413–419.
- Oh, D.Y.; Bang, Y.J. HER2-Targeted Therapies—A Role beyond Breast Cancer. Nat. Rev. Clin. Oncol. 2019, 17, 33–48.
- Seo, A.N.; Kwak, Y.; Kim, D.W.; Kang, S.B.; Choe, G.; Kim, W.H.; Lee, H.S. HER2 Status in Colorectal Cancer: Its Clinical Significance and the Relationship between HER2 Gene Amplification and Expression. PLoS ONE 2014, 9, e98528.
- Kavuri, S.M.; Jain, N.; Galimi, F.; Cottino, F.; Leto, S.M.; Migliardi, G.; Searleman, A.C.; Shen, W.; Monsey, J.; Trusolino, L.; et al. HER2 Activating Mutations Are Targets for Colorectal Cancer Treatment. Cancer Discov. 2015, 5, 832–841.
- El-Deiry, W.S.; Vijayvergia, N.; Xiu, J.; Scicchitano, A.; Lim, B.; Yee, N.S.; Harvey, H.A.; Gatalica, Z.; Reddy, S. Molecular Profiling of 6,892 Colorectal Cancer Samples Suggests Different Possible Treatment Options Specific to Metastatic Sites. Cancer Biol. Ther. 2015, 16, 1726–1737.
- Afrǎsânie, V.A.; Marinca, M.V.; Alexa-Stratulat, T.; Gafton, B.; Pǎduraru, M.; Adavidoaiei, A.M.; Miron, L.; Rusu, C. KRAS, NRAS, BRAF, HER2 and Microsatellite Instability in Metastatic Colorectal Cancer-Practical Implications for the Clinician. Radiol. Oncol. 2019, 53, 265–274.
- Styczen, H.; Nagelmeier, I.; Beissbarth, T.; Nietert, M.; Homayounfar, K.; Sprenger, T.; Boczek, U.; Stanek, K.; Kitz, J.; Wolff, H.A.; et al. HER-2 and HER-3 Expression in Liver Metastases of Patients with Colorectal Cancer. Oncotarget 2015, 6, 15065–15076.
- Fusco, N.; Bosari, S. HER2 Aberrations and Heterogeneity in Cancers of the Digestive System: Implications for Pathologists and Gastroenterologists. World J. Gastroenterol. 2016, 22, 7926–7937.
- Ivanova, M.; Venetis, K.; Guerini-Rocco, E.; Bottiglieri, L.; Mastropasqua, M.G.; Garrone, O.; Fusco, N.; Ghidini, M. HER2 in Metastatic Colorectal Cancer: Pathology, Somatic Alterations, and Perspectives for Novel Therapeutic Schemes. Life 2022, 12, 1403.
- Zhang, X.; Powell, K.; Li, L. Breast Cancer Stem Cells: Biomarkers, Identification and Isolation Methods, Regulating Mechanisms, Cellular Origin, and Beyond. Cancers 2020, 12, 3765.
- Jung, D.H.; Bae, Y.J.; Kim, J.H.; Shin, Y.K.; Jeung, H.C. HER2 Regulates Cancer Stem Cell Activities via the Wnt Signaling Pathway in Gastric Cancer Cells. Oncology 2019, 97, 311–318.
- Liang, K.H.; Tso, H.C.; Hung, S.H.; Kuan, I.I.; Lai, J.K.; Ke, F.Y.; Chuang, Y.T.; Liu, I.J.; Wang, Y.P.; Chen, R.H.; et al. Extracellular Domain of EpCAM Enhances Tumor Progression through EGFR Signaling in Colon Cancer Cells. Cancer Lett. 2018, 433, 165–175.
- Eyvazi, S.; Farajnia, S.; Dastmalchi, S.; Kanipour, F.; Zarredar, H.; Bandehpour, M. Antibody Based EpCAM Targeted Therapy of Cancer, Review and Update. Curr. Cancer Drug. Targets 2018, 18, 857–868.
- Gires, O.; Pan, M.; Schinke, H.; Canis, M.; Baeuerle, P.A. Expression and Function of Epithelial Cell Adhesion Molecule EpCAM: Where Are We after 40 Years? Cancer Metastasis Rev. 2020, 39, 969–987.
- Gao, T.; Cen, Q.; Lei, H. A Review on Development of MUC1-Based Cancer Vaccine. Biomed. Pharmacother. 2020, 132, 110888.
- Guo, M.; Luo, B.; Pan, M.; Li, M.; Xu, H.; Zhao, F.; Dou, J. Colorectal Cancer Stem Cell Vaccine with High Expression of MUC1 Serves as a Novel Prophylactic Vaccine for Colorectal Cancer. Int. Immunopharmacol. 2020, 88, 106850.
- Li, W.; Zhang, N.; Jin, C.; Long, M.D.; Rajabi, H.; Yasumizu, Y.; Fushimi, A.; Yamashita, N.; Hagiwara, M.; Zheng, R.; et al. MUC1-C Drives Stemness in Progression of Colitis to Colorectal Cancer. JCI Insight 2020, 5, e137112.
- Guo, M.; Luo, B.; Pan, M.; Li, M.; Zhao, F.; Dou, J. MUC1 Plays an Essential Role in Tumor Immunity of Colorectal Cancer Stem Cell Vaccine. Int. Immunopharmacol. 2020, 85, 106631.
- Guo, M.; You, C.; Dong, W.; Luo, B.; Wu, Y.; Chen, Y.; Li, J.; Pan, M.; Li, M.; Zhao, F.; et al. The Surface Dominant Antigen MUC1 Is Required for Colorectal Cancer Stem Cell Vaccine to Exert Anti-Tumor Efficacy. Biomed. Pharmacother. 2020, 132, 110804.
- Carrell, R.K.; Stanton, R.A.; Ethier, S.P.; LaRue, A.C.; Soloff, A.C. ICOSL-Augmented Adenoviral-Based Vaccination Induces a Bipolar Th17/Th1 T Cell Response against Unglycosylated MUC1 Antigen. Vaccine 2018, 36, 6262–6269.
- Jones, A.B.; Rocco, A.; Lamb, L.S.; Friedman, G.K.; Hjelmeland, A.B. Regulation of NKG2D Stress Ligands and Its Relevance in Cancer Progression. Cancers 2022, 14, 2339.
- Daniels, R.A.; Turley, H.; Kimberley, F.C.; Liu, X.S.; Mongkolsapaya, J.; Ch’En, P.; Xu, X.N.; Jin, B.Q.; Pezzella, F.; Screaton, G.R. Expression of TRAIL and TRAIL Receptors in Normal and Malignant Tissues. Cell. Res. 2005, 15, 430–438.
- Snajdauf, M.; Havlova, K.; Vachtenheim, J.; Ozaniak, A.; Lischke, R.; Bartunkova, J.; Smrz, D.; Strizova, Z. The TRAIL in the Treatment of Human Cancer: An Update on Clinical Trials. Front. Mol. Biosci. 2021, 8, 628332.
- Kikuchi, Y.; Kunita, A.; Iwata, C.; Komura, D.; Nishiyama, T.; Shimazu, K.; Takeshita, K.; Shibahara, J.; Kii, I.; Morishita, Y.; et al. The Niche Component Periostin Is Produced by Cancer-Associated Fibroblasts, Supporting Growth of Gastric Cancer through ERK Activation. Am. J. Pathol. 2014, 184, 859–870.
- Akbari, M.; Shomali, N.; Faraji, A.; Shanehbandi, D.; Asadi, M.; Mokhtarzadeh, A.; Shabani, A.; Baradaran, B. CD133: An Emerging Prognostic Factor and Therapeutic Target in Colorectal Cancer. Cell Biol. Int. 2020, 44, 368–380.
- Abbasian, M.; Mousavi, E.; Arab-Bafrani, Z.; Sahebkar, A. The Most Reliable Surface Marker for the Identification of Colorectal Cancer Stem-like Cells: A Systematic Review and Meta-Analysis. J. Cell. Physiol. 2019, 234, 8192–8202.
- Ieta, K.; Tanaka, F.; Haraguchi, N.; Kita, Y.; Sakashita, H.; Mimori, K.; Matsumoto, T.; Inoue, H.; Kuwano, H.; Mori, M. Biological and Genetic Characteristics of Tumor-Initiating Cells in Colon Cancer. Ann. Surg. Oncol. 2008, 15, 638–648.
- Vaiopoulos, A.G.; Kostakis, I.D.; Koutsilieris, M.; Papavassiliou, A.G. Colorectal Cancer Stem Cells. Stem Cells 2012, 30, 363–371.
- Todaro, M.; Francipane, M.G.; Medema, J.P.; Stassi, G. Colon Cancer Stem Cells: Promise of Targeted Therapy. Gastroenterology 2010, 138, 2151–2162.
- Zhi, Y.; Mou, Z.; Chen, J.; He, Y.; Dong, H.; Fu, X.; Wu, Y. B7H1 Expression and Epithelial-To-Mesenchymal Transition Phenotypes on Colorectal Cancer Stem-Like Cells. PLoS ONE 2015, 10, e0135528.
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An Embryonic Stem Cell–like Gene Expression Signature in Poorly Differentiated Aggressive Human Tumors. Nat. Genet. 2008, 40, 499–507.
- Schmohl, J.U.; Gleason, M.K.; Dougherty, P.R.; Miller, J.S.; Vallera, D.A. Heterodimeric Bispecific Single Chain Variable Fragments (ScFv) Killer Engagers (BiKEs) Enhance NK-Cell Activity Against CD133+ Colorectal Cancer Cells. Target. Oncol. 2016, 11, 353–361.
- Barzegar Behrooz, A.; Syahir, A.; Ahmad, S. CD133: Beyond a Cancer Stem Cell Biomarker. J. Drug Target. 2018, 27, 257–269.
- Juat, D.J.; Hachey, S.J.; Billimek, J.; Del Rosario, M.P.; Nelson, E.L.; Hughes, C.C.W.; Zell, J.A. Adoptive T-Cell Therapy in Advanced Colorectal Cancer: A Systematic Review. Oncologist 2022, 27, 210–219.
- Ramutton, T.; Buccheri, S.; Dieli, F.; Todaro, M.; Stassi, G.; Meraviglia, S. Γδ T Cells as a Potential Tool in Colon Cancer Immunotherapy. Immunotherapy 2014, 6, 989–999.
- Frank, M.H.; Wilson, B.J.; Gold, J.S.; Frank, N.Y. Clinical Implications of Colorectal Cancer Stem Cells in the Age of Single-Cell Omics and Targeted Therapies. Gastroenterology 2021, 160, 1947–1960.
- Yoshida, Y.; Naito, M.; Yamada, T.; Aisu, N.; Daibo, K.; Mera, T.; Tanaka, T.; Naito, K.; Yasumoto, K.; Kamigaki, T.; et al. Adoptive Chemoimmunotherapy Using Activated T Cells for Stage IV Colorectal Cancer. Anticancer. Res. 2016, 36, 3741–3746.
- Parkhurst, M.R.; Joo, J.; Riley, J.P.; Yu, Z.; Li, Y.; Robbins, P.F.; Rosenberg, S.A. Characterization of Genetically Modified T-Cell Receptors That Recognize the CEA:691-699 Peptide in the Context of HLA-A2.1 on Human Colorectal Cancer Cells. Clin. Cancer Res. 2009, 15, 169–180.
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol. Ther. 2011, 19, 620–626.
- Aparicio, C.; Belver, M.; Enríquez, L.; Espeso, F.; Núñez, L.; Sánchez, A.; de la Fuente, M.Á.; González-vallinas, M. Cell Therapy for Colorectal Cancer: The Promise of Chimeric Antigen Receptor (CAR)-T Cells. Int. J. Mol. Sci. 2021, 22, 1781.
- Li, H.; Yang, C.; Cheng, H.; Huang, S.; Zheng, Y. CAR-T Cells for Colorectal Cancer: Target-Selection and Strategies for Improved Activity and Safety. J. Cancer 2021, 12, 1804–1814.
- Feng, M.; Zhao, Z.; Yang, M.; Ji, J.; Zhu, D. T-Cell Based Immunotherapy in Colorectal Cancer. Cancer Lett. 2020, 498, 201–209.
- Xu, J.; Meng, Q.; Sun, H.; Zhang, X.; Yun, J.; Li, B.; Wu, S.; Li, X.; Yang, H.; Zhu, H.; et al. HER2-Specific Chimeric Antigen Receptor-T Cells for Targeted Therapy of Metastatic Colorectal Cancer. Cell Death Dis. 2021, 12, 1109.
- Magee, M.S.; Abraham, T.S.; Baybutt, T.R.; FlickingerJr, J.C.; Ridge, N.A.; Marszalowicz, G.P.; Prajapati, P.; Hersperger, A.R.; Waldman, S.A.; Snook, A.E. Human GUCY2C-Targeted Chimeric Antigen Receptor (CAR)-Expressing T Cells Eliminate Colorectal Cancer Metastases. Cancer Immunol. Res. 2018, 6, 509–516.
- Abbas, A.K.; Lichtman, A.H.; Pober, J.S. Cellular and Molecular Immunology; W.B. Saunders Company: Philadelphia, PA, USA, 1997.
- Gri, G.; Showe, L. Cutting Edge: Differentiation of Human NK Cells into NK1 and NK2 Subsets. J. Immunol. 1998, 161, 5821–5824.
- Chu, J.; Gao, F.; Yan, M.; Zhao, S.; Yan, Z.; Shi, B.; Liu, Y. Natural Killer Cells: A Promising Immunotherapy for Cancer. J. Transl. Med. 2022, 20, 240.
- Portillo, A.L.; Hogg, R.; Poznanski, S.M.; Rojas, E.A.; Cashell, N.J.; Hammill, J.A.; Chew, M.V.; Shenouda, M.M.; Ritchie, T.M.; Cao, Q.T.; et al. Expanded Human NK Cells Armed with CAR Uncouple Potent Anti-Tumor Activity from off-Tumor Toxicity against Solid Tumors. iScience 2021, 24, 102619.
- Rettinger, E.; KuçI, S.; Naumann, I.; Becker, P.; Kreyenberg, H.; Anzaghe, M.; Willasch, A.; Koehl, U.; Bug, G.; Ruthardt, M.; et al. The Cytotoxic Potential of Interleukin-15-Stimulated Cytokine-Induced Killer Cells against Leukemia Cells. Cytotherapy 2012, 14, 91–103.
- Wang, Y.; Zheng, X.; Wei, H.; Sun, R.; Tian, Z. Different Roles of IL-15 from IL-2 in Differentiation and Activation of Human CD3+CD56+ NKT-like Cells from Cord Blood in Long Term Culture. Int. Immunopharmacol. 2008, 8, 927–934.
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK Cells: Surface Receptors, Inhibitory Checkpoints, and Translational Applications. Cell. Mol. Immunol. 2019, 16, 430–441.
- Shokouhifar, A.; Firouzi, J.; Nouri, M.; Sarab, G.A.; Ebrahimi, M. NK Cell Upraise in the Dark World of Cancer Stem Cells. Cancer Cell Int. 2021, 21, 682.
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting Natural Killer Cells in Cancer Immunotherapy. Nat. Immunol. 2016, 17, 1025–1036.
- Liu, S.; Galat, V.; Galat4, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK Cell-Based Cancer Immunotherapy: From Basic Biology to Clinical Development. J. Hematol. Oncol. 2021, 14, 7.
- Nieda, M. TRAIL Expression by Activated Human CD4+Valpha24NKT Cells Induces in Vitro and in Vivo Apoptosis of Human Acute Myeloid Leukemia Cells. Blood 2001, 97, 2067–2074.
- Grudzien, M.; Rapak, A. Effect of Natural Compounds on NK Cell Activation. J. Immunol. Res. 2018, 2018, 4868417.
- González, B.; Bueno, D.; Rubio, P.M.; Román, S.S.; Plaza, D.; Sastre, A.; García-Miguel, P.; Fernández, L.; Valentín, J.; Martínez, I.; et al. Aspectos Inmunológicos de La Leucemia Mieloblástica Aguda. An. Pediatr. 2016, 84, 195–202.
- Ruggeri, L.; Mancusi, A.; Burchielli, E.; Aversa, F.; Martelli, M.F.; Velardi, A. Natural Killer Cell Alloreactivity and Haplo-Identical Hematopoietic Transplantation. Cytotherapy 2006, 8, 554–558.
- Della Chiesa, M.; Setti, C.; Giordano, C.; Obino, V.; Greppi, M.; Pesce, S.; Marcenaro, E.; Rutigliani, M.; Provinciali, N.; Paleari, L.; et al. NK Cell-Based Immunotherapy in Colorectal Cancer. Vaccines 2022, 10, 1033.
- Wooster, A.L.; Girgis, L.H.; Brazeale, H.; Anderson, T.S.; Wood, L.M.; Lowe, D.B. Dendritic Cell Vaccine Therapy for Colorectal Cancer. Pharmacol. Res. 2021, 164, 105374.
- Kajihara, M.; Takakura, K.; Kanai, T.; Ito, Z.; Saito, K.; Takami, S.; Shimodaira, S.; Okamoto, M.; Ohkusa, T.; Koido, S. Dendritic Cell-Based Cancer Immunotherapy for Colorectal Cancer. World J. Gastroenterol. 2016, 22, 4275.
This entry is offline, you can click here to edit this entry!