The increasing prevalence of obesity (body mass index [BMI] ≥ 25 kg/m
2) has also become an important public health concern [
3,
4]. The worldwide prevalence of obesity increased dramatically by 27.5% for adults between 1980 and 2013 [
3]. Overweight and obesity not only increase the risk of developing BC but also are associated with worse survival compared to patients with normal weight [
4,
5]. BMI is used as a surrogate of abnormal or excessive fat accumulation; however, it is not a reliable measure of body adiposity as BMI does not consider the percentages of lean and fat tissue mass [
6]. Furthermore, the same BMI may correspond to different lean and body fat mass that change with sex, ethnicity and age [
4]. Therefore, further research is required to understand how body composition and BC interact.
Western lifestyle is associated with low-grade inflammation and chronic metabolic inflammatory diseases linked to decreased life expectancy [
7]. Risk factors such as Western diet, reduced physical activity, environmental or socioeconomic factors such as smoking, income, education and occupation, can result in chronic systemic inflammation that activates local immune cells, including macrophages, either directly or indirectly mediated via obesity [
7,
8,
9]. In the context of nutrient excess, adipocytes increase in number and size, which results in adipocyte disruption and cell death [
10]. The innate immune cells, such as macrophages, respond to environmental danger signals that are released by the necrotic adipocytes that trigger chronic low-grade systematic inflammation via metabolic reprogramming [
11]. During inflammation, there is an interplay between macrophages, which act as antigen-presenting cells, and adaptive immunity cells such as T-cells, by shaping T-cell responses and dysregulating the immunometabolic homeostasis towards a proinflammatory environment [
12].
2. Immunometabolic Changes in Obese Adipose Tissue
Under normal physiological conditions, in adipose tissue of individuals with healthy BMI, there is homeostasis between anti-inflammatory and proinflammatory molecules that maintains adipose tissue functions [
15]. Adipose tissue is classified into white and brown adipose tissue (WAT and BAT, respectively) which differ functionally and morphologically. WAT, distributed subcutaneously and viscerally, constitutes 20% of the body weight and 80% of total adipose tissue of a normal adult [
16]. WAT is the largest store of energy, whilst BAT plays a key role in thermogenesis [
16]. Under conditions of nutritional excess and during development of obesity, adipocytes undergo structural alterations, such as adipocyte hypertrophy [
17]. Then adipocytes become dysfunctional, undergo cell death and secrete cytokines that contribute to adipose tissue inflammation and recruitment of pre-adipocytes, leading to adipose tissue hyperplasia [
18,
19]. Apart from structural changes, adipose tissue also undergoes functional changes such as mitochondrial dysfunction and endoplasmic reticulum stress [
20]. In obesity, there is a significant reduction in mitochondrial gene expression leading to downregulation of mitochondrial biogenesis in subcutaneous tissue associated with insulin resistance and inflammation in obese monozygotic twins compared with their leaner co-twins [
21]. Furthermore, free fatty acid-mediated generation of reactive oxygen species is correlated with endoplasmic reticulum stress and upregulation of pro-inflammatory gene signatures in adipose tissue [
22,
23]. Overall, these changes in the adipose tissue result in the activation of proinflammatory signalling pathways, leading to chronic low-grade adipose tissue inflammation mediated by macrophage infiltration, neovascularisation and increase in extracellular matrix [
24,
25,
26,
27] (
Table 1). BC arises in an adipose rich environment and therefore its tumour microenvironment could be impacted by these factors.
Table 1. Obesity is associated with systemic immunometabolic changes.
3. Innate Immunity in Obese Adipose Tissue
During weight gain, adipocytes increase the storage of lipids, resulting in structural changes, such as adipocyte hypertrophy, and adipocyte death. The mechanism of adipocyte death is still not clear, although it has been attributed to either inflammatory programmed cell death (pyroptosis) or necrosis [
38,
39]. Adipose tissue macrophages (ATMs) scavenge the debris of the necrotic adipocytes, which in turn activate ATMs via the initiation of inflammatory signalling pathways [
40,
41,
42,
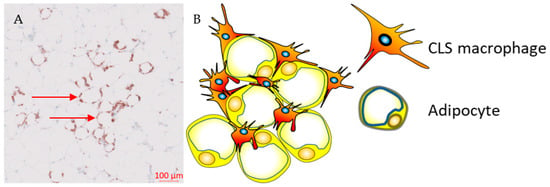
43]. ATMs that are metabolically activated by fatty acids or inflammatory mediators that are released from necrotic adipocytes are recruited or proliferate in situ, and encircle adipocytes forming crown-like structures (CLSs) (
Figure 1) [
44,
45].
Figure 1. Crown-like structures. (A) Representative immunohistochemistry image from the Southampton BEGIN (Investigating outcomes from breast cancer: Correlating genetic, immunological, and nutritional predictors) cohort showing CD68+ crown-like structures within the adipose tissue adjacent to a human breast tumour; (B) hypertrophic adipocytes surrounded by macrophages forming crown-like structures.
Obesity was reported to modulate the phenotype of ATMs from an M2- to M1-like phenotype in mice that received a high fat diet [
46]. ATMs in obese adipose tissue express M2-like markers such as CD163 or CD206, and activate Fcγ receptors such as CD16, as well as a range of M1-like markers, including CD11c, which is involved in T-cell activation in adipose tissue [
45,
47]. This CD11c+ CD163+ subset of ATMs is associated with high BMI and accumulates in the adipose tissue of obese subjects [
48]. CD11c+ CD206+ ATMs confer pro-inflammatory properties that correlate with increased presence of CLS and insulin resistance in obese individuals [
49]. This suggests that ATMs and consequently CLSs are a diverse immune cell population defined by concurrent expression of biomarkers for M1- and M2-like macrophages that is dependent on the presence of adipose tissue in obese people and most likely driven by metabolic dysfunction [
40,
49].
4. Adaptive Immunity in Obese Adipose Tissue
During the development of obesity, the composition of adaptive immune cells resident in adipose tissue changes, with an increase in the CD8+ to CD4+ T-cell ratio, but a reduction in the number of regulatory T-cells (Tregs) within the adipose tissue [
50]. Both CD4+ and CD8+ T-cells play a vital role in the recruitment and activation of ATM via secretion of cytokines such as IFN-γ [
29]. Flow-cytometric and immunohistochemical analyses demonstrated higher numbers of CD8+ effector T-cells and lower numbers of CD4+ helper T-cells in obese murine epididymal adipose tissue compared to lean mice on a normal diet [
29]. In addition, CD8+ T-cells were found within CLS in obese epididymal adipose tissue whereas there was no association was found between CD4+ T-cells and CLS [
29]. A time course evaluation of immune cells in adipose tissue in C57BL/6 mice during a high-fat diet showed that CD8+ T-cell infiltration preceded the recruitment of macrophages [
29]. In contrast, the number of CD4+CD8- helper T-cells and CD4+CD25+FoxP3+ Tregs decreased, suggesting that CD8+ T-cell infiltration is a crucial event during inflammation in adipose tissue [
29]. This was further validated by depleting CD8+ T-cells in C57BL/6 mice using anti-CD8 antibody, which resulted in reduction of M1-like macrophages and CLSs without affecting M2-like macrophages [
29]. In addition, high-fat diet did not increase levels of IL-6 and TNF-α mRNA in CD8-deficient mice, whereas adoptive transfer of CD8+ T-cells into CD8-deficient mice increased M1-like macrophage infiltration [
29].
These findings suggest that Tregs maintain immune homeostasis by suppressing inflammation induced by pro-inflammatory macrophages in adipose tissue under physiological conditions. CD8+ T-cell infiltration is required for adipose tissue inflammation in obesity as it precedes macrophage accumulation in adipose tissue and plays a vital role in macrophage polarisation and infiltration. Obesity-induced metabolic dysregulation may interfere in the interplay between macrophages and T-cell immune populations [
29]. Differences in the immunophenotype of ATMs between non-obese and obese subjects may be attributed to their different immunometabolic functions influenced by metabolic stress and chronic inflammation, which is promoted by enlarged or necrotic adipocytes.
5. Adipose Tissue Macrophages and Breast Cancer
CLSs are correlated with a proinflammatory environment and represent an index of WAT inflammation and metabolic dysregulation such as dyslipidemia, increased glucose and glycated hemoglobin (HbA1) levels [
44,
45,
51,
52]. Observational studies in patients with early breast cancer demonstrated that chronic systemic inflammation, as defined by elevated serum proinflammatory cytokines, is associated with CLSs, especially in individuals with obesity or who are overweight [
51,
53,
54]. Previous reports have demonstrated an inconsistency in survival, with three out of five studies reporting improved outcomes (
Table 2), which can be partly explained by biological and methodological heterogeneity [
45,
51,
55,
56]. Our recent study showed that the presence of CLSs expressing the inhibitory FcγRIIB (CD32B) at the tumour border was associated with worse clinical outcomes in patients with HER2+ breast cancer treated with trastuzumab compared to trastuzumab-naïve patients [
45]. The underlying biological mechanism that links the presence of CD32B+ CLS and resistance to trastuzumab is currently unclear and further investigation is required.
Table 2. Clinical outcomes of retrospective cohorts that evaluated the prognostic role of CLSs in breast cancer.
Griner et al. investigated the role of adipocytes in trastuzumab resistance in HER2+ BT474 and SKBR-3 cell lines. Culture of these cell lines with conditioned media from in vitro differentiated adipocytes was associated with improved viability and AKT phosphorylation in the trastuzumab-treated HER2+ cell lines compared to the controls [
60]. Pharmacological blockade of PI3K via the PI3K inhibitor LY294002 or transfection with an inactive AKT1 kinase mutant reversed the resistance to trastuzumab that was induced by the conditioned media [
60]. It was also reported that leptin enhances the overexpression of HER2 receptor and cell proliferation of HER2+ breast cancer cell lines, which results in resistance to tamoxifen [
61,
62]. Leptin-induced overexpression of the HER2 receptor is mediated via the activation of the RAS-dependent MAPK pathway, which phosphorylates both epidermal growth factor receptor and Janus-activated kinase 2 [
61,
62]. Stimulation of the MCF7 cell line with leptin was associated with HER2 phosphorylation on Tyr1248, which led to the activation of proliferation and survival pathways [
63]. Leptin-dependent activation of the HER2 receptor could be explained by the co-localisation of leptin and HER2 receptors in HER2+ cell lines and human breast tumours [
63].
In ER+ BC, aromatase expression and activity are associated with both high BMI and WAT inflammation [
64]. Interestingly, WAT inflammation, as defined by the presence of CLSs, is associated with raised aromatase expression and activity in women with normal BMI [
54,
65]. Furthermore, adipocyte size and markers of subclinical systemic inflammation are strongly associated with increased levels of aromatase in postmenopausal women [
64]. In addition, menopause is associated with a reduction in oestrogen levels, which is linked to the development of obesity [
66]. Treatment with 17β-oestradiol protected ovariectomised mice against high fat diet induced weight gain and was associated with a reduction in aromatase expression, WAT inflammation and the associated proinflammatory mediators in the mammary glands, which are mediated via oestrogen receptor-α [
66]. Hence, administration of oestrogen in obese mice may modulate WAT inflammation either through weight loss or due to its potential anti-inflammatory properties [
66].
The role of adiposity and inflammation was also demonstrated in ER− BC. A systematic review and meta-analysis that included 13 observational studies of patients with triple negative BC (TNBC) with baseline BMI measurements, showed that BMI ≥ 25 was associated with worse disease-free and overall survival compared to patients with healthy BMI [74]. In a study of 1779 patients with primary invasive BC, patients with triple-negative disease had a 3-fold risk of being overweight and of having raised CRP compared to luminal A subjects [70]. Recent studies proposed potential mechanisms underlying the association between obesity and TNBC. These include the activation of Akt/mTOR signalling pathway by insulin, which is elevated in patients with obesity-induced insulin resistance [76]. Activation of the Akt/mTOR pathway is associated with aggressive molecular and glycolytic phenotypes that promote tumour growth in TNBC [77,78]. Secondly, obesity-mediated inflammation has been associated with activation-signalling pathways that are involved in tumour invasion and metastasis in TNBC [79]. Thirdly, this chronic inflammatory environment was reported to be correlated with reduced tumour-infiltrating immune cells both in a 4T1 TNBC model and in human triple negative tumours [80].
These findings suggest that hyperadiposity can induce WAT inflammation and metabolic dysregulation. In ER+ breast tumours, this is potentially mediated via a paracrine interaction between macrophages and preadipocytes, leading to elevated aromatase expression and secretion of pro-inflammatory adipokines in the breast adipose tissue in patients with high BMI [
44]. In ER− BC obesity is positively associated with inflammatory and aggressive molecular phenotypes. In contrast, in HER2+ breast tumours, WAT inflammation can induce trastuzumab resistance via activation of MAPK or PI3K pathways [
60].