Skin inflammation is a common underlying feature of atopic dermatitis, allergic contact dermatitis and chronic spontaneous urticaria. miRNAs are involved in the pathogenesis and regulation of atopic dermatitis and can reveal an atopic predisposition or indicate disease severity. In chronic spontaneous urticaria, different miRNAs which are over-expressed during urticaria exacerbations not only play a role in the possible response to therapy or remission, but also serve as a marker of chronic autoimmune urticaria and indicate associations with other autoimmune diseases. In allergic contact dermatitis, miRNAs are upregulated in inflammatory lesions and expressed during the sensitization phase of allergic response. Several miRNAs have been identified as potential biomarkers of these chronic skin conditions, but they are also possible therapeutic targets.
- miRNA
- atopic dermatitis
- chronic spontaneous urticaria
- allergic contact dermatitis
- antagomirs
- cytokines
- inflammation
- skin
- pathogenesis
- treatment
1. Introduction
1.1. Pathogenesis of Atopic Dermatitis
1.2. Pathogenesis of Allergic Contact Dermatitis
1.3. Pathogenesis of Chronic Spontaneous Urticaria
1.4. MicroRNAs
2. miRNA in AD, ACD and CSU: Pathogenetic Role and Therapeutic Strategies
2.1. Pro-Inflammatory and Anti-Inflammatory miRNA in AD
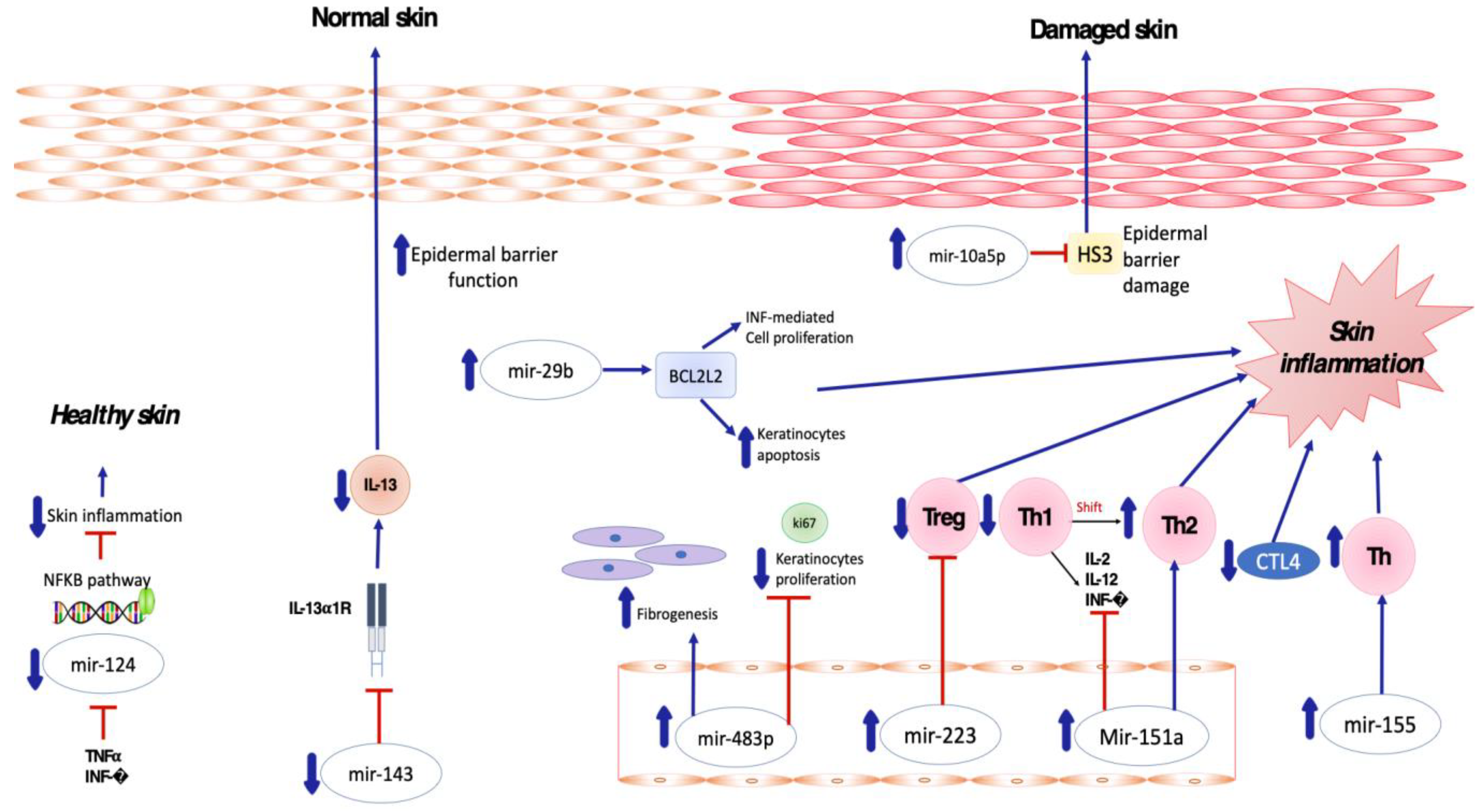
2.2. Pro-Inflammatory and Anti-Inflammatory miRNA in ACD
2.3. Pro-Inflammatory and Anti-Inflammatory miRNA in CSU
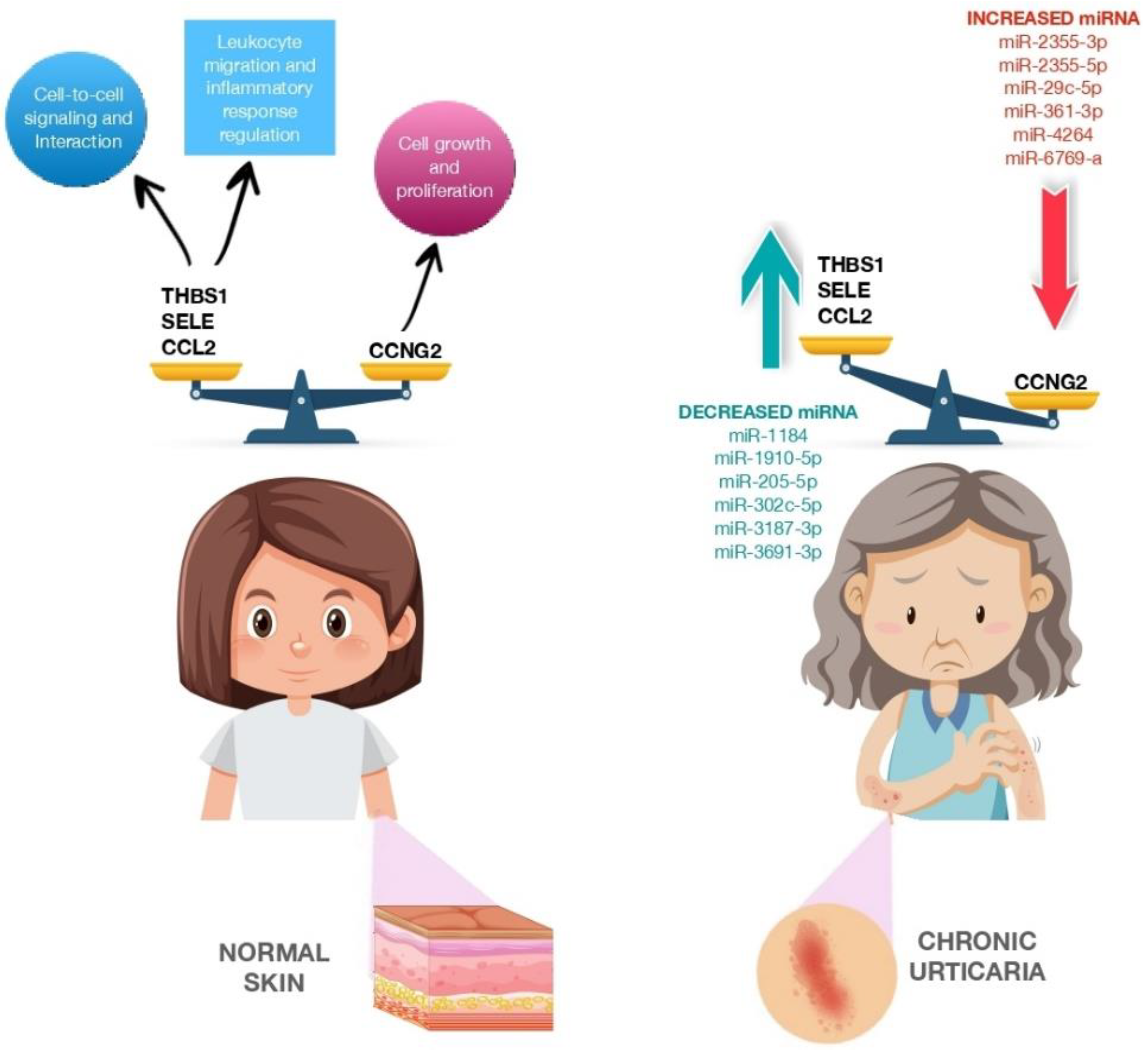
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11051266
References
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic Dermatitis. Nat. Rev. Dis. Primers 2018, 4, 1.
- Nettis, E.; Patella, V.; Brancaccio, R.; Detoraki, C.; Di Leo, E.; Incorvaia, C.; Macchia, L.; Pellacani, G.; Bonzano, L. Efficacy of Dupilumab in Concomitant Atopic Dermatitis and Chronic Rhinosinusitis With Nasal Polyps: A Preliminary Study. Allergy Asthma Immunol. Res. 2021, 13, 347–349.
- Megna, M.; Patruno, C.; Balato, A.; Rongioletti, F.; Stingeni, L.; Balato, N.; Ayala, F.; Brambilla, L.; Congedo, M.; Corazza, M.; et al. An Italian Multicentre Study on Adult Atopic Dermatitis: Persistent versus Adult-Onset Disease. Arch. Derm. Res. 2017, 309, 443–452.
- Kraft, M.; Worm, M. Dupilumab in the Treatment of Moderate-to-Severe Atopic Dermatitis. Expert Rev. Clin. Immunol. 2017, 13, 301–310.
- Patel, N.; Feldman, S.R. Management of Atopic Dermatitis. Adherence in Atopic Dermatitis. Introduction. Adv. Exp. Med. Biol. 2017, 1027, 139–159.
- Elias, P.M.; Steinhoff, M. “Outside-to-inside” (and Now Back to “Outside”) Pathogenic Mechanisms in Atopic Dermatitis. J. Investig. Dermatol. 2008, 128, 1067–1070.
- Bertino, L.; Guarneri, F.; Cannavò, S.P.; Casciaro, M.; Pioggia, G.; Gangemi, S. Oxidative Stress and Atopic Dermatitis. Antioxidants 2020, 9, 196.
- Nedoszytko, B.; Reszka, E.; Gutowska-Owsiak, D.; Trzeciak, M.; Lange, M.; Jarczak, J.; Niedoszytko, M.; Jablonska, E.; Romantowski, J.; Strapagiel, D.; et al. Genetic and Epigenetic Aspects of Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 6484.
- Zaniboni, M.C.; Samorano, L.P.; Orfali, R.L.; Aoki, V. Skin Barrier in Atopic Dermatitis: Beyond Filaggrin. Bras Derm. 2016, 91, 472–478.
- Katsunuma, T.; Kawahara, H.; Yuki, K.; Akasawa, A.; Saito, H. Impaired Interferon-Gamma Production in a Subset Population of Severe Atopic Dermatitis. Int. Arch. Allergy Immunol. 2004, 134, 240–247.
- Malik, K.; Heitmiller, K.D.; Czarnowicki, T. An Update on the Pathophysiology of Atopic Dermatitis. Derm. Clin. 2017, 35, 317–326.
- Palmer, C.N.A.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.D.; et al. Common Loss-of-Function Variants of the Epidermal Barrier Protein Filaggrin Are a Major Predisposing Factor for Atopic Dermatitis. Nat. Genet. 2006, 38, 441–446.
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.J.; et al. A Role for IL-25 and IL-33-Driven Type-2 Innate Lymphoid Cells in Atopic Dermatitis. J. Exp. Med. 2013, 210, 2939–2950.
- Ito, T.; Wang, Y.H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.F.; Yao, Z.; Cao, W.; Liu, Y.J. TSLP-Activated Dendritic Cells Induce an Inflammatory T Helper Type 2 Cell Response through OX40 Ligand. J. Exp. Med. 2005, 202, 1213–1223.
- Gandhi, N.A.; Bennett, B.L.; Graham, N.M.H.; Pirozzi, G.; Stahl, N.; Yancopoulos, G.D. Targeting Key Proximal Drivers of Type 2 Inflammation in Disease. Nat. Rev. Drug Discov. 2016, 15, 35–50.
- Islam, S.A.; Luster, A.D. T Cell Homing to Epithelial Barriers in Allergic Disease. Nat. Med. 2012, 18, 705–715.
- Liu, B.; Tai, Y.; Achanta, S.; Kaelberer, M.M.; Caceres, A.I.; Shao, X.; Fang, J.; Jordt, S.E. IL-33/ST2 Signaling Excites Sensory Neurons and Mediates Itch Response in a Mouse Model of Poison Ivy Contact Allergy. Proc. Natl. Acad. Sci. USA 2016, 113, E7572–E7579.
- Nassau, S.; Fonacier, L. Allergic Contact Dermatitis. Med. Clin. N. Am. 2020, 104, 61–76.
- Zack, B.; Arrandale, V.H.; Holness, D.L. Preventing Occupational Skin Disease: A Review of Training Programs. Dermatitis 2017, 28, 169–182.
- Rustemeyer, T.; Van Hoogstraten, I.M.W.; Von Blomberg, B.M.E.; Gibbs, S.; Scheper, R.J. Mechanisms of Irritant and Allergic Contact Dermatitis. Contact Dermat. 2011, 43–90.
- Adelman, D.C.; Casale, T.B.; Corren, J.; Ovid Technologies, Inc. Manual of Allergy and Immunology; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; p. 494.
- Zuberbier, T.; Abdul Latiff, A.H.; Abuzakouk, M.; Aquilina, S.; Asero, R.; Baker, D.; Ballmer-Weber, B.; Bangert, C.; Ben-Shoshan, M.; Bernstein, J.A.; et al. The International EAACI/GA2LEN/EuroGuiDerm/APAAACI Guideline for the Definition, Classification, Diagnosis, and Management of Urticaria. Allergy Eur. J. Allergy Clin. Immunol. 2022, 77, 734–766.
- Nettis, E.; Cegolon, L.; Di Leo, E.; Lodi Rizzini, F.; Detoraki, A.; Canonica, G.W. Omalizumab in Chronic Spontaneous Urticaria: Efficacy, Safety, Predictors of Treatment Outcome, and Time to Response. Ann. Allergy Asthma Immunol. 2018, 121, 474–478.
- Vestergaard, C.; Toubi, E.; Maurer, M.; Triggiani, M.; Ballmer-Weber, B.; Marsland, A.; Ferrer, M.; Knulst, A.; Giménez-Arnau, A. Treatment of Chronic Spontaneous Urticaria with an Inadequate Response to H1-Antihistamines: An Expert Opinion. Eur. J. Dermatol. 2017, 27, 10–19.
- Bracken, S.J.; Abraham, S.; MacLeod, A.S. Autoimmune Theories of Chronic Spontaneous Urticaria. Front. Immunol. 2019, 10, 627.
- Grattan, C.E.H.; Dawn, G.; Gibbs, S.; Francis, D.M. Blood Basophil Numbers in Chronic Ordinary Urticaria and Healthy Controls: Diurnal Variation, Influence of Loratadine and Prednisolone and Relationship to Disease Activity. Clin. Exp. Allergy 2003, 33, 337–341.
- Kern, F.; Lichtenstein, L.M. Defective Histamine Release in Chronic Urticaria. J. Clin. Investig. 1976, 57, 1369–1377.
- Sun, R.S.; Sui, J.F.; Chen, X.H.; Ran, X.Z.; Yang, Z.F.; Guan, W.D.; Yang, T. Detection of CD4+ CD25+ FOXP3+ Regulatory T Cells in Peripheral Blood of Patients with Chronic Autoimmune Urticaria. Australas J. Dermatol. 2011, 52, e15–e18.
- Zhu, Y.; Huang, Y.; Ming, B.; Wu, X.; Chen, Y.; Dong, L. Regulatory T-Cell Levels in Systemic Lupus Erythematosus Patients: A Meta-Analysis. Lupus 2019, 28, 445–454.
- Morita, T.; Shima, Y.; Wing, J.B.; Sakaguchi, S.; Ogata, A.; Kumanogoh, A. The Proportion of Regulatory T Cells in Patients with Rheumatoid Arthritis: A Meta-Analysis. PLoS ONE 2016, 11, e0162306.
- Vasagar, K.; Vonakis, B.M.; Gober, L.M.; Viksman, A.; Gibbons, S.P.; Saini, S.S. Evidence of in Vivo Basophil Activation in Chronic Idiopathic Urticaria. Clin. Exp. Allergy 2006, 36, 770–776.
- Ulambayar, B.; Chen, Y.H.; Ban, G.Y.; Lee, J.H.; Jung, C.G.; Yang, E.M.; Park, H.S.; Ye, Y.M. Detection of Circulating IgG Autoantibody to FcεRIα in Sera from Chronic Spontaneous Urticaria Patients. J. Microbiol. Immunol. Infect. 2020, 53, 141–147.
- Zhang, L.; Qi, R.; Yang, Y.; Gao, X.; Chen, H.; Xiao, T. Serum MiR-125a-5p and CCL17 Upregulated in Chronic Spontaneous Urticaria and Correlated with Treatment Response. Acta Derm. Venereol. 2019, 99, 571–578.
- Ambros, V.; Lee, R.C.; Lavanway, A.; Williams, P.T.; Jewell, D. MicroRNAs and Other Tiny Endogenous RNAs in C. Elegans. Curr. Biol. 2003, 13, 807–818.
- Setoyama, T.; Ling, H.; Natsugoe, S.; Calin, G.A. Non-Coding RNAs for Medical Practice in Oncology. Keio J. Med. 2011, 60, 106–113.
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most Mammalian MRNAs Are Conserved Targets of MicroRNAs. Genome Res. 2009, 19, 92–105.
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA Polymerase III Transcribes Human MicroRNAs. Nat. Struct Mol. Biol. 2006, 13, 1097–1101.
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of the Sequence and Temporal Expression of Let-7 Heterochronic Regulatory RNA. Nature 2000, 408, 86–89.
- Hashimoto, Y.; Akiyama, Y.; Yuasa, Y. Multiple-to-multiple relationships between microRNAs and target genes in gastric cancer. PLoS ONE 2013, 8, e62589.
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating High Confidence MicroRNAs Using Deep Sequencing Data. Nucleic. Acids Res. 2014, 42, D68–D73.
- Sayed, D.; Abdellatif, M. MicroRNAs in Development and Disease. Physiol. Rev. 2011, 91, 827–887.
- Hutvágner, G.; Zamore, P.D. A MicroRNA in a Multiple-Turnover RNAi Enzyme Complex. Science 2002, 297, 2056–2060.
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates That Thousands of Human Genes Are MicroRNA Targets. Cell 2005, 120, 15–20.
- Hu, H.Y.; Yan, Z.; Xu, Y.; Hu, H.; Menzel, C.; Zhou, Y.H.; Chen, W.; Khaitovich, P. Sequence Features Associated with MicroRNA Strand Selection in Humans and Flies. BMC Genom. 2009, 10, 413.
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of MRNA Translation and Stability by MicroRNAs. Annu. Rev. Biochem. 2010, 79, 351–379.
- Zhou, W.Y.; Cai, Z.R.; Liu, J.; Wang, D.S.; Ju, H.Q.; Xu, R.H. Circular RNA: Metabolism, functions and interactions with proteins. Mol. Cancer 2020, 14, 172.
- Tong, K.L.; Tan, K.E.; Lim, Y.Y.; Tien, X.Y.; Wong, P.F. CircRNA-miRNA interactions in atherogenesis. Mol. Cell. Biochem. 2022, 477, 2703–2733.
- Huntzinger, E.; Izaurralde, E. Gene Silencing by MicroRNAs: Contributions of Translational Repression and MRNA Decay. Nat. Rev. Genet. 2011, 12, 99–110.
- Ambros, V. The Functions of Animal MicroRNAs. Nature 2004, 431, 350–355.
- Fiorucci, G.; Chiantore, M.V.; Mangino, G.; Percario, Z.A.; Affabris, E.; Romeo, G. Cancer Regulator MicroRNA: Potential Relevance in Diagnosis, Prognosis and Treatment of Cancer. Curr. Med. Chem. 2012, 19, 461–474.
- Shelburne, C.P.; Ryan, J.J. The Role of Th2 Cytokines in Mast Cell Homeostasis. Immunol. Rev. 2001, 179, 82–93.
- Banerjee, J.; Sen, C.K. MicroRNAs in skin and wound healing. Methods Mol. Biol. 2013, 936, 343–356.
- Frankel, H.C.; Qureshi, A.A. Comparative Effectiveness of Topical Calcineurin Inhibitors in Adult Patients with Atopic Dermatitis. Am. J. Clin. Dermatol. 2012, 13, 113–123.
- Sonkoly, E.; Janson, P.; Majuri, M.L.; Savinko, T.; Fyhrquist, N.; Eidsmo, L.; Xu, N.; Meisgen, F.; Wei, T.; Bradley, M.; et al. MiR-155 Is Overexpressed in Patients with Atopic Dermatitis and Modulates T-Cell Proliferative Responses by Targeting Cytotoxic T Lymphocyte-Associated Antigen 4. J. Allergy Clin. Immunol. 2010, 126, 581–589.e20.
- Sonkoly, E.; Pivarcsi, A. Advances in MicroRNAs: Implications for Immunity and Inflammatory Diseases. J. Cell Mol. Med. 2009, 13, 24–38.
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; Van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of Bic/MicroRNA-155 for Normal Immune Function. Science 2007, 316, 608–611.
- Thai, T.H.; Calado, D.P.; Casola, S.; Ansel, K.M.; Xiao, C.; Xue, Y.; Murphy, A.; Frendewey, D.; Valenzuela, D.; Kutok, J.L.; et al. Regulation of the Germinal Center Response by MicroRNA-155. Science 2007, 316, 604–608.
- Rebane, A.; Runnel, T.; Aab, A.; Maslovskaja, J.; Rückert, B.; Zimmermann, M.; Plaas, M.; Kärner, J.; Treis, A.; Pihlap, M.; et al. MicroRNA-146a Alleviates Chronic Skin Inflammation in Atopic Dermatitis through Suppression of Innate Immune Responses in Keratinocytes. J. Allergy Clin. Immunol. 2014, 134, 836–847.e11.
- West, C.; McDermott, M.F. Effects of MicroRNA-146a on the Proliferation and Apoptosis of Human Osteochondrocytes by Targeting TRAF6 through the NF- ΚB Signalling Pathway. Biosci. Rep. 2017, 37, 180.
- Park, H.; Huang, X.; Lu, C.; Cairo, M.S.; Zhou, X. MicroRNA-146a and MicroRNA-146b Regulate Human Dendritic Cell Apoptosis and Cytokine Production by Targeting TRAF6 and IRAK1 Proteins. J. Biol. Chem. 2015, 290, 2831–2841.
- Lindner, J.M.; Kayo, H.; Hedlund, S.; Fukuda, Y.; Fukao, T.; Nielsen, P.J. Cutting Edge: The Transcription Factor Bob1 Counteracts B Cell Activation and Regulates MiR-146a in B Cells. J. Immunol. 2014, 192, 4483–4486.
- Williams, A.E.; Perry, M.M.; Moschos, S.A.; Larner-Svensson, H.M.; Lindsay, M.A. Role of MiRNA-146a in the Regulation of the Innate Immune Response and Cancer. Biochem. Soc. Trans. 2008, 36, 1211–1215.
- Chen, X.F.; Zhang, L.J.; Zhang, J.; Dou, X.; Shao, Y.; Jia, X.J.; Zhang, W.; Yu, B. MiR-151a Is Involved in the Pathogenesis of Atopic Dermatitis by Regulating Interleukin-12 Receptor Β2. Exp. Dermatol. 2018, 27, 427–432.
- Kim, C.H.; Park, C.D.; Lee, A.Y. Administration of Poly(I:C) Improved Dermatophagoides Farinae-Induced Atopic Dermatitis-like Skin Lesions in NC/Nga Mice by the Regulation of Th1/Th2 Balance. Vaccine 2012, 30, 2405–2410.
- Guo, H.W.; Yun, C.X.; Hou, G.H.; Du, J.; Huang, X.; Lu, Y.; Keller, E.T.; Zhang, J.; Deng, J.G. Mangiferin Attenuates TH1/TH2 Cytokine Imbalance in an Ovalbumin-Induced Asthmatic Mouse Model. PLoS ONE 2014, 9, e100394.
- Zeng, Y.P.; Nguyen, G.H.; Jin, H.Z. MicroRNA-143 Inhibits IL-13-Induced Dysregulation of the Epidermal Barrier-Related Proteins in Skin Keratinocytes via Targeting to IL-13Rα1. Mol. Cell Biochem. 2016, 416, 63–70.
- Jeong, D.; Kim, J.; Nam, J.; Sun, H.; Lee, Y.H.; Lee, T.J.; Aguiar, R.C.T.; Kim, S.W. MicroRNA-124 Links P53 to the NF-ΚB Pathway in B-Cell Lymphomas. Leukemia 2015, 29, 1868–1874.
- Yang, Z.; Zeng, B.; Wang, C.; Wang, H.; Huang, P.; Pan, Y. MicroRNA-124 Alleviates Chronic Skin Inflammation in Atopic Eczema via Suppressing Innate Immune Responses in Keratinocytes. Cell Immunol. 2017, 319, 53–60.
- Vennegaard, M.T.; Bonefeld, C.M.; Hagedorn, P.H.; Bangsgaard, N.; Løvendorf, M.B.; Odum, N.; Woetmann, A.; Geisler, C.; Skov, L. Allergic Contact Dermatitis Induces Upregulation of Identical MicroRNAs in Humans and Mice. Contact Dermat. 2012, 67, 298–305.
- Gulati, N.; Løvendorf, M.B.; Zibert, J.R.; Akat, K.M.; Renwick, N.; Tuschl, T.; Krueger, J.G. Unique MicroRNAs Appear at Different Times during the Course of a Delayed-Type Hypersensitivity Reaction in Human Skin. Exp. Dermatol. 2015, 24, 953–957.
- Anderson, S.E.; Beezhold, K.; Lukomska, E.; Richardson, J.; Long, C.; Anderson, K.; Franko, J.; Meade, B.J.; Beezhold, D.H. Expression Kinetics of MiRNA Involved in Dermal Toluene 2,4-Diisocyanate Sensitization. J. Immunotoxicol. 2014, 11, 250–259.
- Werner, P.; Wisgrill, L.; Riskumäki, M.; Jalonen, E.; Vendelin, J.; Suomela, S.; Lauerma, A.; Alenius, H.; Fyhrquist, N. Identification of Novel MiRNA-MRNA Regulatory Networks in Contact Dermatitis by Integrated Microarray Analysis. Allergy 2021, 76, 1257–1261.
- Lin, C.K.E.; Kaptein, J.S.; Sheikh, J. Differential Expression of MicroRNAs and Their Possible Roles in Patients with Chronic Idiopathic Urticaria and Active Hives. Allergy Rhinol. 2017, 8, e67–e80.