Cranial and peripheral nerve sheath tumors (PNST) comprise a heterogeneous group of soft tissue tumors. Most arise from classic peripheral nervous system elements (Schwann cells and perineurial cells), while others involve specialized neuroendocrine cells of the sympathetic and parasympathetic nervous system (e.g., cauda equina neuroendocrine tumors, previously known as “CNS paragangliomas”). The World Health Organization (WHO) classification of the Central Nervous System (CNS) Tumors of 2021 and the 2020 WHO Classification of Soft Tissue and Bone Tumors include benign and malignant tumors, such as schwannoma, neurofibroma, plexiform neurofibroma (PN), perineurioma, hybrid nerve sheath tumor (HNST), malignant peripheral nerve sheath tumor (MPNST), epithelioid MPNST or malignant melanotic nerve sheath tumor (MMNST), and cauda equine neuroendocrine tumor. All these entities may arise along the craniospinal axis and be encountered either sporadically or as part of neurocutaneous syndromes, including neurofibromatosis type 1 (NF1), schwannoma predisposition syndromes (SPS), and Carney complex.
- schwannoma
- neurofibroma
- plexiform neurofibroma
- perineurioma
1. Epidemiology and Clinical Features
2. Genetic Tumor Syndromes Correlated with Cranial and Peripheral Nerve Sheath Tumors
3. Pathology and Molecular Markers
Tumor diagnosis for most of cranial and paraspinal nerve sheath tumors is still primarily based on hematoxylin and eosin (H&E)-stained sections and some additional techniques, including immunohistochemistry. Molecular testing generally is not required for this type of tumor but may be of help in the distinction of low-grade MPNST from cellular or atypical neurofibroma. However, mutation analysis of PNST may be required to diagnose mosaic forms of NF1, NF2, and SPS through the identification of the same mutation in at least two independent tumors as this is often the only way to prove a mosaicism.
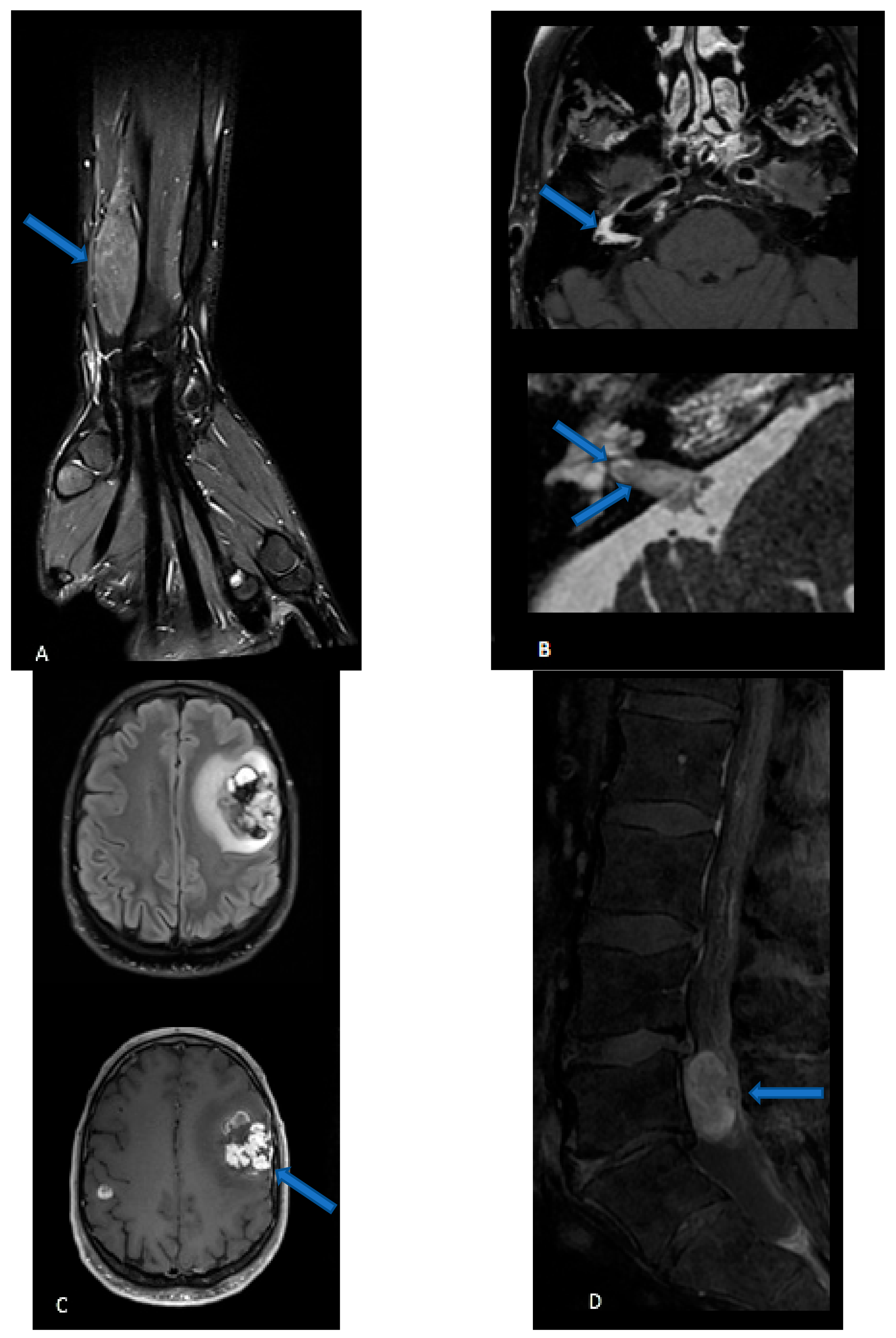
4. Imaging
5. Surgery
6. Radiotherapy
7. Medical Treatments
7.1. Vestibular Schwannomas
7.2. Plexiform Neurofibromas
7.3. Malignant Nerve Sheath Tumors
8. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/cancers15071930
References
- Brainin, M.; Barnes, M.; Baron, J.C.; Gilhus, N.E.; Hughes, R.; Selmaj, K.; Waldemar, G.; Guideline Standards Subcommittee of the EFNS Scientific Committee. Guidance for the preparation of neurological management guidelines by EFNS scientific task forces—Revised recommendations 2004. Eur. J. Neurol. 2004, 11, 577–581.
- Casadei, G.P.; Komori, T.; Scheithauer, B.W.; Miller, G.M.; Parisi, J.E.; Kelly, P.J. Intracranial parenchymal schwannoma. A clinico-pathological and neuroimaging study of nine cases. J. Neurosurg. 1993, 79, 217–222.
- Voltaggio, L.; Murray, R.; Lasota, J.; Miettinen, M. Gastric schwannoma: A clinicopathologic study of 51 cases and critical review of the literature. Hum. Pathol. 2012, 43, 650–659.
- Babu, R.; Sharma, R.; Bagley, J.H.; Hatef, J.; Friedman, A.H.; Adamson, C. Vestibular schwannomas in the modern era: Epidemiology, treatment trends, and disparities in management. J. Neurosurg. 2013, 119, 121–130.
- Evans, D.G.R. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J. Rare Dis. 2009, 4, 16.
- Andersen, J.F.; Nilsen, K.S.; Vassbotn, F.S.; Møller, P.; Myrseth, E.; Lund-Johansen, M.; Goplen, F.K. Predictors of Vertigo in Patients with Untreated Vestibular Schwannoma. Otol. Neurotol. 2015, 36, 647–652.
- Woodruff, J.M.; Selig, A.M.; Crowley, K.; Allen, P.W. Schwannoma (Neurilemoma) with Malignant Transformation A Rare, Distinctive Peripheral Nerve Tumor. Am. J. Surg. Pathol. 1994, 18, 882–895.
- McMenamin, M.E.; Fletcher, C.D.M. Expanding the Spectrum of Malignant Change in Schwannomas: Epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: A study of 17 cases. Am. J. Surg. Pathol. 2001, 25, 13–25.
- Carter, J.M.; O’Hara, C.; Dundas, G.; Gilchrist, D.; Collins, M.S.; Eaton, K.; Judkins, A.R.; Biegel, J.A.; Folpe, A.L. Epithelioid Malignant Peripheral Nerve Sheath Tumor Arising in a Schwannoma, in a Patient With “Neuroblastoma-like” Schwannomatosis and a Novel Germline SMARCB1 Mutation. Am. J. Surg. Pathol. 2012, 36, 154–160.
- Perry, A.; Reuss, D.E.; Rodriguez, F. Neurofibroma. In WHO Classification of Tumours Series, 5th ed.; IARC Press: Lyon, France, 2020; Volume 3, pp. 232–236.
- Wolkenstein, P.; Zeller, J.; Revuz, J.; Ecosse, E.; Leplège, A. Quality-of-Life Impairment in Neurofibromatosis Type 1: A cross-sectional study of 128 cases. Arch. Dermatol. 2001, 137, 1421–1425.
- Meyer, A.; Billings, S.D. What’s new in nerve sheath tumors. Virchows Arch. 2020, 476, 65–80.
- Collins-Sawaragi, Y.C.; Ferner, R.; Vassallo, G.; De Agrò, G.; Eccles, S.; Cadwgan, J.; Hargrave, D.; Hupton, E.; Eelloo, J.; Lunt, L.; et al. Location, symptoms, and management of plexiform neurofibromas in 127 children with neurofibromatosis 1, attending the National Complex Neurofibromatosis 1 service, 2018–2019. Am. J. Med. Genet. Part A 2022, 188, 1723–1727.
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1—A consensus overview. Hum. Pathol. 2017, 67, 1–10.
- Bs, J.E.B.; Peterson, C.R.; Dhakal, S.; Giampoli, E.J.; Constine, L.S. Malignant peripheral nerve sheath tumors (MPNST): A SEER analysis of incidence across the age spectrum and therapeutic interventions in the pediatric population. Pediatr. Blood Cancer 2014, 61, 1955–1960.
- Somatilaka, B.N.; Sadek, A.; McKay, R.M.; Le, L.Q. Malignant peripheral nerve sheath tumor: Models, biology, and translation. Oncogene 2022, 41, 2405–2421.
- Ferner, R.E.; Gutmann, D.H. International consensus statement on malignant peripheral nerve sheath tumors in neurofibroma-tosis. Cancer Res. 2002, 62, 1573–1577.
- Evans, D.G.R.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314.
- Foley, K.M.; Woodruff, J.M.; Ellis, F.T.; Posner, J.B. Radiation-induced malignant and atypical peripheral nerve sheath tumors. Ann. Neurol. 1980, 7, 311–318.
- Stewart, D.R.; Korf, B.R.; Nathanson, K.L.; Stevenson, D.A.; Yohay, K. Care of adults with neurofibromatosis type 1: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Anesth. Analg. 2018, 20, 671–682.
- Carli, M.; Ferrari, A.; Mattke, A.; Zanetti, I.; Casanova, M.; Bisogno, G.; Cecchetto, G.; Alaggio, R.; De Sio, L.; Koscielniak, E.; et al. Pediatric Malignant Peripheral Nerve Sheath Tumor: The Italian and German Soft Tissue Sarcoma Cooperative Group. J. Clin. Oncol. 2005, 23, 8422–8430.
- Wong, W.W.; Hirose, T.; Scheithauer, B.W.; Schild, S.; Gunderson, L.L. Malignant peripheral nerve sheath tumor: Analysis of treatment outcome. Int. J. Radiat. Oncol. 1998, 42, 351–360.
- Stucky, C.-C.H.; Johnson, K.N.; Gray, R.J.; Pockaj, B.A.; Ocal, I.T.; Rose, P.S.; Wasif, N. Malignant Peripheral Nerve Sheath Tumors (MPNST): The Mayo Clinic Experience. Ann. Surg. Oncol. 2011, 19, 878–885.
- Valentin, T.; Le Cesne, A.; Ray-Coquard, I.; Italiano, A.; Decanter, G.; Bompas, E.; Isambert, N.; Thariat, J.; Linassier, C.; Bertucci, F.; et al. Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO). Eur. J. Cancer 2016, 56, 77–84.
- Torres-Mora, J.; Dry, S.; Li, X.; Binder, S.; Amin, M.; Folpe, A.L. Malignant Melanotic Schwannian Tumor: A clinicopathologic, im-munohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of “melanotic schwannoma”. Am. J. Surg. Pathol. 2014, 38, 94–105.
- Carney, J.A. Psammomatous melanotic schwannoma. A distinctive, heritable tumor with special associations, including cardiac myxoma and the Cushing syndrome. Am. J. Surg. Pathol. 1990, 14, 206–222.
- Zhang, H.-Y.; Yang, G.-H.; Chen, H.-J.; Wei, B.; Ke, Q.; Guo, H.; Ye, L.; Bu, H.; Yang, K.; Zhang, Y.-H. Clinicopathological, immunohistochemical, and ultrastructural study of 13 cases of melanotic schwannoma. Chin. Med. J. 2005, 118, 1451–1461.
- Wang, L.; Zehir, A.; Sadowska, J.; Zhou, N.; Rosenblum, M.; Busam, K.; Agaram, N.; Travis, W.; Arcila, M.; Dogan, S.; et al. Consistent copy number changes and recurrentPRKAR1Amutations distinguish Melanotic Schwannomas from Melanomas: SNP-array and next generation sequencing analysis. Genes Chromosom. Cancer 2015, 54, 463–471.
- Lenartowicz, K.A.; Goyal, A.; Mauermann, M.L.; Wilson, T.J.; Spinner, R.J. Clinical Features, Natural History, and Outcomes of Intraneural Perineuriomas: A Systematic Review of the Literature. World Neurosurg. 2021, 154, 120–131.e8.
- Almefty, R.; Webber, B.L.; Arnautović, K.I. Intraneural perineurioma of the third cranial nerve: Occurrence and identification: Case report. J. Neurosurg. 2006, 104, 824–827.
- Christoforidis, M.; Buhl, R.; Paulus, W.; Sepehrnia, A. intraneural perineurioma of the viiith cranial nerve: Case report. Neurosurgery 2007, 61, E652.
- Giannini, C.; Scheithauer, B.W.; Steinberg, J.; Cosgrove, T.J. Intraventricular Perineurioma: Case Report. Neurosurgery 1998, 43, 1478–1481.
- Mauermann, M.L.; Amrami, K.K.; Kuntz, N.L.; Spinner, R.J.; Dyck, P.J.; Bosch, E.P.; Engelstad, J.; Felmlee, J.P.; Dyck, P.J. Longitudinal study of intraneural perineurioma--a benign, focal hypertrophic neuropathy of youth. Brain 2009, 132, 2265–2276.
- Hornick, J.L.; Bundock, E.A.; Fletcher, C.D.M. Hybrid Schwannoma/Perineurioma: Clinicopathologic analysis of 42 distinctive benign nerve sheath tumors. Am. J. Surg. Pathol. 2009, 33, 1554–1561.
- Michal, M.; Kazakov, D.V.; Belousova, I.; Bisceglia, M.; Zamecnik, M.; Mukensnabl, P. A benign neoplasm with histopathological features of both schwannoma and retiform perineurioma (benign schwannoma-perineurioma): A report of six cases of a distinctive soft tissue tumor with a predilection for the fingers. Virchows Arch. 2004, 445, 347–353.
- Goyal-Honavar, A.; Gupta, A.; Chacko, G.; Chacko, A.G. Trigeminal hybrid nerve sheath tumor—A case report and literature review. Br. J. Neurosurg. 2021, 1–4.
- Las Heras, F.L.; Martuza, R.; Caruso, P.; Rincon, S.; Stemmer-Rachamimov, A. 24-Year-Old Woman with An Internal Auditory Canal Mass. Hybrid peripheral nerve sheath tumor with schwannoma/perineurioma components. Brain Pathol. 2013, 23, 361–362.
- Matsuo, S.; Higaki, R.; Matsukado, K. Microsurgical Resection of Hybrid Nerve Sheath Tumor Involving the Orbit and Lateral Wall of the Cavernous Sinus: 2-Dimensional Operative Video. Oper. Neurosurg. 2021, 21, E551.
- Harder, A.; Wesemann, M.; Hagel, C.; Schittenhelm, J.; Fischer, S.; Tatagiba, M.; Nagel, C.; Jeibmann, A.; Bohring, A.; Mautner, V.-F.; et al. Hybrid Neurofibroma/Schwannoma is Overrepresented Among Schwannomatosis and Neurofibromatosis Patients. Am. J. Surg. Pathol. 2012, 36, 702–709.
- MacCollin, M.; Chiocca, E.A.; Evans, G.; Friedman, J.; Horvitz, R.; Jaramillo, D.; Lev, M.; Mautner, V.F.; Niimura, M.; Plotkin, S.R.; et al. Diagnostic criteria for schwannomatosis. Neurology 2005, 64, 1838–1845.
- Kacerovska, D.; Michal, M.; Kuroda, N.; Tanaka, A.; Sima, R.; Denisjuk, N.; Kreuzberg, B.; Ricarova, R.; Kazakov, D.V. Hybrid Peripheral Nerve Sheath Tumors, Including a Malignant Variant in Type 1 Neurofibromatosis. Am. J. Dermatopathol. 2013, 35, 641–649.
- Inatomi, Y.; Ito, T.; Nagae, K.; Yamada, Y.; Kiyomatsu, M.; Nakano-Nakamura, M.; Uchi, H.; Oda, Y.; Furue, M. Hybrid perineurioma-neurofibroma in a patient with neurofibromatosis type 1, clinically mimicking malignant peripheral nerve sheath tumor. Eur. J. Dermatol. 2014, 24, 412–413.
- Easton, D.F.; Ponder, M.A.; Huson, S.M.; Ponder, B.A. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): Evidence for modifying genes. Am. J. Hum. Genet. 1993, 53, 305–313.
- Jordan, J.T.; Plotkin, S.R. Neurofibromatoses. Hematol. Oncol. Clin. N. Am. 2022, 36, 253–267.
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Anesth. Analg. 2021, 23, 1506–1513.
- Messiaen, L. Multidiscipilinary Approach to Neurofibromatosis 1. In Molecular Diagnosis of NF1; Tadini, G., Legius, E., Brems, H., Eds.; Springer: Cham, Switzerland, 2020.
- Plotkin, S.R.; Messiaen, L.; Legius, E.; Pancza, P.; Avery, R.A.; Blakeley, J.O.; Babovic-Vuksanovic, D.; Ferner, R.; Fisher, M.J.; Friedman, J.M.; et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Anesth. Analg. 2022, 24, 1967–1977.
- Smith, M.J.; Bowers, N.L.; Banks, C.; Coates-Brown, R.; Morris, K.A.; Ewans, L.; Wilson, M.; Pinner, J.; Bhaskar, S.S.; Cammarata-Scalisi, F.; et al. A deep intronic SMARCB1 variant associated with schwannomatosis. Clin. Genet. 2020, 97, 376–377.
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251.
- The WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Lyon, France, 2020.
- Nonaka, D.; Chiriboga, L.; Rubin, B.P. Sox10: A Pan-Schwannian and Melanocytic Marker. Am. J. Surg. Pathol. 2008, 32, 1291–1298.
- Karamchandani, J.R.; Nielsen, T.O.; van de Rijn, M.; West, R.B. Sox10 and S100 in the Diagnosis of Soft-tissue Neoplasms. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 445–450.
- Jo, V.Y.; Fletcher, C.D. SMARCB1/INI1 Loss in Epithelioid Schwannoma: A Clinicopathologic and Immunohistochemical Study of 65 Cases. Am. J. Surg. Pathol. 2017, 41, 1013–1022.
- Schaefer, I.-M.; Dong, F.; Garcia, E.P.; Fletcher, C.D.M.; Jo, V.Y. Recurrent SMARCB1 Inactivation in Epithelioid Malignant Peripheral Nerve Sheath Tumors. Am. J. Surg. Pathol. 2019, 43, 835–843.
- Colman, S.D.; Williams, C.A.; Wallace, M.R. Benign neurofibromas in type 1 neurofibromatosis (NF1) show somatic deletions of the NF1 gene. Nat. Genet. 1995, 11, 90–92.
- Klein, C.J.; Wu, Y.; Jentoft, M.E.; Mer, G.; Spinner, R.J.; Dyck, P.J.B.; Mauermann, M.L. Genomic analysis reveals frequentTRAF7mutations in intraneural perineuriomas. Ann. Neurol. 2017, 81, 316–321.
- Carter, J.M.; Wu, Y.; Blessing, M.M.; Folpe, A.L.; Thorland, E.C.; Spinner, R.J.; Jentoft, M.E.; Wang, C.; Baheti, S.; Niu, Z.; et al. Recurrent Genomic Alterations in Soft Tissue Perineuriomas. Am. J. Surg. Pathol. 2018, 42, 1708–1714.
- Brock, J.E.; Perez-Atayde, A.R.; Kozakewich, H.P.W.; Richkind, K.E.; Fletcher, J.A.; Vargas, S.O. Cytogenetic Aberrations in Perineurioma: Variation with subtype. Am. J. Surg. Pathol. 2005, 29, 1164–1169.
- Ronellenfitsch, M.W.; Harter, P.N.; Kirchner, M.; Heining, C.; Hutter, B.; Gieldon, L.; Schittenhelm, J.; Schuhmann, M.U.; Tatagiba, M.; Marquardt, G.; et al. Targetable ERBB2 mutations identified in neurofibroma/schwannoma hybrid nerve sheath tumors. J. Clin. Investig. 2020, 130, 2488–2495.
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Granada, C.N.P.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232.
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251.
- Zhang, M.; Wang, Y.; Jones, S.; Sausen, M.; McMahon, K.; Sharma, R.; Wang, Q.; Belzberg, A.J.; Chaichana, K.; Gallia, G.L.; et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1170–1172.
- Pemov, A.; Hansen, N.F.; Sindiri, S.; Patidar, R.; Higham, C.S.; Dombi, E.; Miettinen, M.M.; Fetsch, P.; Brems, H.; Chandrasekharappa, S.C.; et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1–associated atypical neurofibromas. Neuro-Oncology 2019, 21, 981–992.
- Prieto-Granada, C.N.; Wiesner, T.; Messina, J.L.; Jungbluth, A.A.; Chi, P.; Antonescu, C.R. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am. J. Surg. Pathol. 2016, 40, 479–489.
- Schaefer, I.-M.; Fletcher, C.D.; Hornick, J.L. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod. Pathol. 2016, 29, 4–13.
- Walker, F.O.; Cartwright, M.S.; Alter, K.E.; Visser, L.H.; Hobson-Webb, L.D.; Padua, L.; Strakowski, J.A.; Preston, D.C.; Boon, A.J.; Axer, H.; et al. Indications for neuromuscular ultrasound: Expert opinion and review of the literature. Clin. Neurophysiol. 2018, 129, 2658–2679.
- Hung, E.H.Y.; Griffith, J.F.; Ng, A.W.H.; Lee, R.K.L.; Lau, D.T.Y.; Leung, J.C.S. Ultrasound of Musculoskeletal Soft-Tissue Tumors Superficial to the Investing Fascia. Am. J. Roentgenol. 2014, 202, W532–W540.
- Tøttrup, M.; Eriksen, J.D.; Hellfritzsch, M.B.; Sørensen, F.B.; Baad-Hansen, T. Diagnostic accuracy of ultrasound-guided core biopsy of peripheral nerve sheath tumors. J. Clin. Ultrasound 2020, 48, 134–138.
- Pendleton, C.; Spinner, R.J. Image-guided percutaneous biopsy of peripheral nerve tumors of indeterminate nature: Risks and benefits. Acta Neurochir. 2020, 162, 1425–1429.
- Grover, S.B.; Kundra, R.; Grover, H.; Gupta, V.; Gupta, R. Imaging diagnosis of plexiform neurofibroma- unravelling the confounding features: A report of two cases. Radiol. Case Rep. 2021, 16, 2824–2833.
- Bruscella, S.; Alfieri, A.; de Bellis, A.; Rolando, F.; Covelli, E.M.; Manfredonia, L.; Orabona, P.; de Marinis, P. Malignant Intracerebral Nerve Sheath Tumor Presenting with Intratumoral Hemorrhage. World Neurosurg. 2021, 145, 370–375.
- Tanaka, M.; Shibui, S.; Nomura, K.; Nakanishi, Y.; Hasegawa, T.; Hirose, T.; Le Fèvre, C.; Castelli, J.; Perrin, C.; Hénaux, P.; et al. Malignant intracerebral nerve sheath tumor with intratumoral calcification. J. Neurosurg. 2000, 92, 338–341.
- Agarwal, A.; Chandra, A.; Jaipal, U.; Bagarhatta, M.; Mendiratta, K.; Goyal, A.; Kumar, R.; Mangalhara, N. Can imaging be the new yardstick for diagnosing peripheral neuropathy?—A comparison between high resolution ultrasound and MR neurography with an approach to diagnosis. Insights Imaging 2019, 10, 104–113.
- Noebauer-Huhmann, I.M.; Weber, M.-A.; Lalam, R.K.; Trattnig, S.; Bohndorf, K.; Vanhoenacker, F.; Tagliafico, A.; Van Rijswijk, C.; Vilanova, J.C.; Afonso, P.D.; et al. Soft Tissue Tumors in Adults: ESSR-Approved Guidelines for Diagnostic Imaging. Semin. Musculoskelet. Radiol. 2015, 19, 475–482.
- Laffan, E.E.; Ngan, B.-Y.; Navarro, O.M. Pediatric Soft-Tissue Tumors and Pseudotumors: MR Imaging Features with Pathologic Correlation: Part 2. Tumors of Fibroblastic/Myofibroblastic, So-called Fibrohistiocytic, Muscular, Lymphomatous, Neurogenic, Hair Matrix, and Uncertain Origin. Radiographics 2009, 29, e36.
- Ahlawat, S.; Chhabra, A.; Blakely, J. Magnetic Resonance Neurography of Peripheral Nerve Tumors and Tumorlike Conditions. Neuroimaging Clin. N. Am. 2014, 24, 171–192.
- Fisher, S.; Karri, S.; Ramzi, A.; Sharma, R.; Chhabra, A.; Soldatos, T. Advanced MR Imaging of Peripheral Nerve Sheath Tumors Including Diffusion Imaging. Semin. Musculoskelet. Radiol. 2015, 19, 179–190.
- Chhabra, A.; Deshmukh, S.D.; Lutz, A.M.; Fritz, J.; Andreisek, G.; Sneag, D.B.; Subhawong, T.; Singer, A.D.; Wong, P.K.; Thakur, U.; et al. Neuropathy Score Reporting and Data System: A Reporting Guideline for MRI of Peripheral Neuropathy with a Multicenter Validation Study. Am. J. Roentgenol. 2022, 219, 279–291.
- Chhabra, A.; Deshmukh, S.D.; Lutz, A.M.; Fritz, J.; Sneag, D.B.; Mogharrabi, B.; Guirguis, M.; Andreisek, G.; Xi, Y.; Ahlawat, S. Neuropathy Score Reporting and Data System (NS-RADS): MRI Reporting Guideline of Peripheral Neuropathy Explained and Reviewed. Skelet. Radiol. 2022, 51, 1909–1922.
- Broski, S.M.; Johnson, G.; Howe, B.M.; Nathan, M.A.; Wenger, D.E.; Spinner, R.J.; Amrami, K.K. Evaluation of 18F-FDG PET and MRI in differentiating benign and malignant peripheral nerve sheath tumors. Skelet. Radiol. 2016, 45, 1097–1105.
- Raad, R.A.; Lala, S.; Allen, J.C.; Babb, J.; Mitchell, C.W.; Franceschi, A.M.; Yohay, K.; Friedman, K.P. Comparison of hybrid 18F-fluorodeoxyglucose positron emission tomography/magnetic resonance imaging and positron emission tomography/computed tomography for evaluation of peripheral nerve sheath tumors in patients with neurofibromatosis type 1. World J. Nucl. Med. 2018, 17, 241–248.
- Reinert, C.P.; Schuhmann, M.U.; Bender, B.; Gugel, I.; la Fougère, C.; Schäfer, J.; Gatidis, S. Comprehensive anatomical and functional imaging in patients with type I neurofibromatosis using simultaneous FDG-PET/MRI. Eur. J. Nucl. Med. 2019, 46, 776–787.
- Bredella, M.A.; Torriani, M.; Hornicek, F.; Ouellette, H.A.; Plamer, W.E.; Williams, Z.; Fischman, A.J.; Plotkin, S.R. Value of PET in the Assessment of Patients with Neurofibromatosis Type 1. Am. J. Roentgenol. 2007, 189, 928–935.
- Li, C.-S.; Huang, G.-S.; Wu, H.-D.; Chen, W.-T.; Shih, L.-S.; Lii, J.-M.; Duh, S.-J.; Chen, R.-C.; Tu, H.-Y.; Chan, W.P. Differentiation of soft tissue benign and malignant peripheral nerve sheath tumors with magnetic resonance imaging. Clin. Imaging 2008, 32, 121–127.
- Mautner, V.F.; Hartmann, M.; Kluwe, L.; Friedrich, R.E.; Fünsterer, C. MRI growth patterns of plexiform neurofibromas in patients with neurofibromatosis type 1. Neuroradiology 2006, 48, 160–165.
- Razek, A.A.K.A.; Ashmalla, G.A. Assessment of paraspinal neurogenic tumors with diffusion-weighted MR imaging. Eur. Spine J. 2018, 27, 841–846.
- Fayad, L.M.; Wang, X.; Blakeley, J.O.; Durand, D.J.; Jacobs, M.A.; Demehri, S.; Subhawong, T.K.; Soldatos, T.; Barker, P.B. Characterization of Peripheral Nerve Sheath Tumors with 3T Proton MR Spectroscopy. Am. J. Neuroradiol. 2014, 35, 1035–1041.
- Ogose, A.; Hotta, T.; Morita, T.; Higuchi, T.; Umezu, H.; Imaizumi, S.; Hatano, H.; Kawashima, H.; Gu, W.; Endo, N. Diagnosis of Peripheral Nerve Sheath Tumors around the Pelvis. Jpn. J. Clin. Oncol. 2004, 34, 405–413.
- Lang, S.-S.; Zager, E.L.; Coyne, T.M.; Nangunoori, R.; Kneeland, J.B.; Nathanson, K. Hybrid peripheral nerve sheath tumor: Case report. J. Neurosurg. 2012, 117, 897–901.
- Koeller, K.K.; Shih, R.Y. Intradural Extramedullary Spinal Neoplasms: Radiologic-Pathologic Correlation. RadioGraphics 2019, 39, 468–490.
- Nguyen, R.; Dombi, E.; Widemann, B.C.; Solomon, J.; Fuensterer, C.; Kluwe, L.; Friedman, J.M.; Mautner, V.-F. Growth dynamics of plexiform neurofibromas: A retrospective cohort study of 201 patients with neurofibromatosis 1. Orphanet J. Rare Dis. 2012, 7, 75.
- Rubino, F.; Eichberg, D.G.; Shah, A.H.; Luther, E.M.; Lu, V.M.; Saad, A.G.; Kahn, D.; Komotar, R.J.; Ivan, M.E. When “Peripheral” Becomes “Central”: Primary and Secondary Malignant Intracerebral Nerve Sheath Tumor: A Case Report and a Systematic Review. Neurosurgery 2021, 88, 1074–1087.
- Le Fèvre, C.; Castelli, J.; Perrin, C.; Hénaux, P.L.; Noël, G. Tumeurs malignes des gaines nerveuses périphériques intracérébrales métastatiques: À propos de deux cas et revue exhaustive des cas de la littérature. Cancer/Radiothérapie 2016, 20, 119–132.
- Klekamp, J.; Samii, M. Surgery of Spinal Nerve Sheath Tumors with Special Reference to Neurofibromatosis. Neurosurgery 1998, 42, 279–289.
- Robla-Costales, J.; Rodríguez-Aceves, C.; Martínez-Benia, F.; Socolovsky, M. State of the Art and Advances in Peripheral Nerve Surgery. Adv. Tech. Stand. Neurosurg. 2022, 45, 245–283.
- Zipfel, J.; Al-Hariri, M.; Gugel, I.; Grimm, A.; Steger, V.; Ladurner, R.; Krimmel, M.; Tatagiba, M.; Schuhmann, M. Surgical Management of Sporadic Peripheral Nerve Schwannomas in Adults: Indications and Outcome in a Single Center Cohort. Cancers 2021, 13, 1017.
- Germano, I.M.; Sheehan, J.; Parish, J.; Atkins, T.; Asher, A.; Hadjipanayis, C.G.; Burri, S.H.; Green, S.; Olson, J.J. Congress of Neurological Surgeons Systematic Review and Evidence-Based Guidelines on the Role of Radiosurgery and Radiation Therapy in the Management of Patients with Vestibular Schwannomas. Neurosurgery 2018, 82, E49–E51.
- Paldor, I.; Chen, A.S.; Kaye, A.H. Growth rate of vestibular schwannoma. J. Clin. Neurosci. 2016, 32, 1–8.
- Hunter, J.B.; Francis, D.O.; O’Connell, B.P.; Kabagambe, E.K.; Bennett, M.L.; Wanna, G.B.; Rivas, A.; Thompson, R.C.; Haynes, D.S. Single Institutional Experience with Observing 564 Vestibular Schwannomas: Factors Associated with Tumor Growth. Otol. Neurotol. 2016, 37, 1630–1636.
- Tveiten, O.V.; Carlson, M.L.; Goplen, F.; Vassbotn, F.; Link, M.J.; Lund-Johansen, M. Long-term Auditory Symptoms in Patients with Sporadic Vestibular Schwannoma: An International Cross-Sectional Study. Neurosurgery 2015, 77, 218–227.
- Kahn, J.; Gillespie, A.; Tsokos, M.; Ondos, J.; Dombi, E.; Camphausen, K.; Widemann, B.C.; Kaushal, A. Radiation Therapy in Management of Sporadic and Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors. Front. Oncol. 2014, 4, 324.
- Martin, E.; Coert, J.H.; Flucke, U.E.; Slooff, W.-B.M.; Ho, V.K.; van der Graaf, W.T.; van Dalen, T.; van de Sande, M.A.; van Houdt, W.J.; Grünhagen, D.J.; et al. A nationwide cohort study on treatment and survival in patients with malignant peripheral nerve sheath tumours. Eur. J. Cancer 2020, 124, 77–87.
- Spunt, S.L.; Million, L.; Chi, Y.-Y.; Anderson, J.; Tian, J.; Hibbitts, E.; Coffin, C.; McCarville, M.B.; Randall, R.L.; Parham, D.M.; et al. A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): A Children’s Oncology Group prospective study. Lancet Oncol. 2020, 21, 145–161.
- Plotkin, S.R.; Merker, V.L.; Halpin, C.; Jennings, D.; McKenna, M.J.; Harris, G.J.; Barker, F.G.I., 2nd. Bevacizumab for Progressive Vestibular Schwannoma in Neurofibromatosis Type 2: A retrospective review of 31 patients. Otol. Neurotol. 2012, 33, 1046–1052.
- Blakeley, J.O.; Ye, X.; Duda, D.G.; Halpin, C.F.; Bergner, A.L.; Muzikansky, A.; Merker, V.; Gerstner, E.R.; Fayad, L.M.; Ahlawat, S.; et al. Efficacy and Biomarker Study of Bevacizumab for Hearing Loss Resulting from Neurofibromatosis Type 2–Associated Vestibular Schwannomas. J. Clin. Oncol. 2016, 34, 1669–1675.
- Robertson, K.A.; Nalepa, G.; Yang, F.-C.; Bowers, D.C.; Ho, C.Y.; Hutchins, G.D.; Croop, J.M.; Vik, T.A.; Denne, S.C.; Parada, L.F.; et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: A phase 2 trial. Lancet Oncol. 2012, 13, 1218–1224.
- Widemann, B.C.; Dombi, E.; Gillespie, A.; Wolters, P.L.; Belasco, J.; Goldman, S.; Korf, B.R.; Solomon, J.; Martin, S.; Salzer, W.; et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro-Oncology 2014, 16, 707–718.
- Widemann, B.C.; Babovic-Vuksanovic, D.; Dombi, E.; Wolters, P.L.; Goldman, S.; Martin, S.; Goodwin, A.; Goodspeed, W.; Kieran, M.W.; Cohen, B.; et al. Phase II trial of pirfenidone in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Pediatr. Blood Cancer 2014, 61, 1598–1602.
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.A.; Cantor, A.; Korf, B.; Perentesis, J.; Gutmann, D.H.; Schorry, E.; et al. Sirolimus for non-progressive NF1-associated plexiform neurofibromas: An NF clinical trials consortium phase II study. Pediatr. Blood Cancer 2013, 61, 982–986.
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.; Cantor, A.; Perentesis, J.; Schorry, E.; Ullrich, N.; Gutmann, D.H.; et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: A Neurofibromatosis Clinical Trials Consortium phase II study. Neuro-Oncology 2015, 17, 596–603.
- Jakacki, R.I.; Dombi, E.; Steinberg, S.M.; Goldman, S.; Kieran, M.W.; Ullrich, N.J.; Pollack, I.F.; Goodwin, A.; Manley, P.E.; Fangusaro, J.; et al. Phase II trial of pegylated interferon alfa-2b in young patients with neurofibromatosis type 1 and unresectable plexiform neurofibromas. Neuro-Oncology 2017, 19, 289–297.
- Zehou, O.; Ferkal, S.; Brugieres, P.; Barbarot, S.; Bastuji-Garin, S.; Combemale, P.; Valeyrie-Allanore, L.; Sbidian, E.; Wolkenstein, P. Absence of Efficacy of Everolimus in Neurofibromatosis 1-Related Plexiform Neurofibromas: Results from a Phase 2a Trial. J. Investig. Dermatol. 2019, 139, 718–720.
- Gross, A.M.; Dombi, E.; Widemann, B.C. Current status of MEK inhibitors in the treatment of plexiform neurofibromas. Child’s Nerv. Syst. 2020, 36, 2443–2452.
- Weiss, B.D.; Wolters, P.L.; Plotkin, S.R.; Widemann, B.C.; Tonsgard, J.H.; Blakeley, J.; Allen, J.C.; Schorry, E.; Korf, B.; Robison, N.J.; et al. NF106: A Neurofibromatosis Clinical Trials Consortium Phase II Trial of the MEK Inhibitor Mirdametinib (PD-0325901) in Adolescents and Adults with NF1-Related Plexiform Neurofibromas. J. Clin. Oncol. 2021, 39, 797–806.
- McCowage, G.B.; Mueller, S.; Pratilas, C.A.; Hargrave, D.R.; Moertel, C.L.; Whitlock, J.; Fox, E.; Hingorani, P.; Russo, M.W.; Dasgupta, K.; et al. Trametinib in pediatric patients with neurofibromatosis type 1 (NF-1)–associated plexiform neurofibroma: A phase I/IIa study. J. Clin. Oncol. 2018, 36, 10504.
- Cai, Z.; Tang, X.; Liang, H.; Yang, R.; Yan, T.; Guo, W. Prognosis and risk factors for malignant peripheral nerve sheath tumor: A systematic review and meta-analysis. World J. Surg. Oncol. 2020, 18, 257.
- Yan, P.; Huang, R.; Hu, P.; Liu, F.; Zhu, X.; Hu, P.; Yin, H.; Zhang, J.; Meng, T.; Huang, Z. Nomograms for predicting the overall and cause-specific survival in patients with malignant peripheral nerve sheath tumor: A population-based study. J. Neuro-Oncol. 2019, 143, 495–503.
- Judson, I.; Verweij, J.; Gelderblom, H.; Hartmann, J.T.; Schöffski, P.; Blay, J.-Y.; Kerst, J.M.; Sufliarsky, J.; Whelan, J.; Hohenberger, P.; et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: A randomised controlled phase 3 trial. Lancet Oncol. 2014, 15, 415–423.
- Skotheim, R.I.; Kallioniemi, A.; Bjerkhagen, B.; Mertens, F.; Brekke, H.R.; Monni, O.; Mousses, S.; Mandahl, N.; Sœter, G.; Nesland, J.M.; et al. Topoisomerase-IIα Is Upregulated in Malignant Peripheral Nerve Sheath Tumors and Associated with Clinical Outcome. J. Clin. Oncol. 2003, 21, 4586–4591, Erratum in J. Clin. Oncol. 2004, 22, 969.
- Higham, C.S.; Steinberg, S.M.; Dombi, E.; Perry, A.; Helman, L.J.; Schuetze, S.M.; Ludwig, J.A.; Staddon, A.; Milhem, M.; Rushing, D.; et al. SARC006: Phase II Trial of Chemotherapy in Sporadic and Neurofibromatosis Type 1 Associated Chemotherapy-Naive Malignant Peripheral Nerve Sheath Tumors. Sarcoma 2017, 2017, 8685638.
- Steins, M.B.; Serve, H.; Zühlsdorf, M.; Senninger, N.; Semik, M.; Berdel, W.E. Carboplatin/etoposide induces remission of metasta-sised malignant peripheral nerve tumours (malignant schwannoma) refractory to first-line therapy. Oncol Rep. 2002, 9, 627–630.
- Leu, K.M.; Ostruszka, L.J.; Shewach, D.; Zalupski, M.; Sondak, V.; Biermann, J.S.; Lee, J.S.-J.; Couwlier, C.; Palazzolo, K.; Baker, L.H. Laboratory and Clinical Evidence of Synergistic Cytotoxicity of Sequential Treament with Gemcitabine Followed by Docetaxel in the Treatment of Sarcoma. J. Clin. Oncol. 2004, 22, 1706–1712.
- Takahashi, M.; Komine, K.; Imai, H.; Okada, Y.; Saijo, K.; Takahashi, M.; Shirota, H.; Ohori, H.; Takahashi, S.; Chiba, N.; et al. Efficacy and safety of gemcitabine plus docetaxel in Japanese patients with unresectable or recurrent bone and soft tissue sarcoma: Results from a single-institutional analysis. PLoS ONE 2017, 12, e0176972.
- Moretti, V.M.; Crawford, E.A.; Staddon, A.P.; Lackman, R.D.; Ogilvie, C.M. Early Outcomes for Malignant Peripheral Nerve Sheath Tumor Treated with Chemotherapy. Am. J. Clin. Oncol. 2011, 34, 417–421.