Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Primary localized cutaneous nodular amyloidosis (PLCNA) is a rare condition attributed to plasma cell proliferation and the deposition of immunoglobulin light chains in the skin without association with systemic amyloidosis or hematological dyscrasias. It is not uncommon for patients diagnosed with PLCNA to also suffer from other auto-immune connective tissue diseases, with Sjögren’s syndrome (SjS) showing the strongest association.
- localized cutaneous nodular amyloidosis
- plasma cells
- Sjögren’s Syndrome
- auto-immune connective tissue disorders
1. Primary Localized Cutaneous Nodular Amyloidosis: A Small Subset of the Large Group of Cutaneous Amyloidosis
The amyloid substance is a fibrillar proteinaceous material composed of two components: a common one derived from the serum amyloid component and a specific one that defines the type of amyloidosis [1]. Amyloidosis can affect both the skin and internal organs. Cutaneous manifestations are not exclusive to the localized cutaneous type but can also occur in systemic forms of amyloidosis, which may be primary due to plasma cell dyscrasia or secondary due to chronic inflammatory disorders. Primary localized cutaneous amyloidosis can be divided into three forms, the two most common being lichenoid amyloidosis (LA) and macular amyloidosis (MA). They are characterized by amyloid deposition in the upper dermis. In PLCNA, the third and least common form, the amyloid, affects the dermis, subcutaneous tissue, and blood vessels and is usually accompanied by plasma cell infiltrates [2].
MA and LA may represent a single clinical spectrum. These two conditions have an epidermal origin, as the genesis of amyloid deposits is the degeneration of basal keratinocytes. This process is often precipitated by the chronic rubbing of or friction to the skin. Cytokeratins, which are the target of auto-antibodies, are released during the apoptosis of keratinocytes, and cytokeratin 5 (also 1, 10, and 14) is the major component of amyloid. After subsequent phagocytosis by macrophages, the resulting enzyme product becomes amyloid K. Clinically, MA presents as hyperpigmented, thin plaques, often with “wavy” linear grayish-brown streaks. The scapular region and extensor areas of the extremities are the most frequently affected areas. In contrast, LA is manifested as discrete, millimeter-sized skin-to-hyperpigmented colored dome-shaped papules. These papules overlap to form plaques, with the calves being a typical site. A rare variant known as “biphasic cutaneous amyloidosis” has been described, in which lichenoid and macular lesions occur at the same time [3].
Unlike the other two forms of primary localized amyloidosis, the amyloid in PLCNA is derived from immunoglobulin light chains (amyloid AL) (either κ or λ) and, thus, from plasma cells. However, an isolated case of PLCNA has been described in which the amyloid substance was found to be of the K-type rather than the AL-type [4]. Although, in systemic amyloidosis, the amyloid substance is also derived from immunoglobulin light chains, whereas in PLCNA, the plasma cells are found only in the skin and not in the bone marrow since it is not associated with an underlying hematologic dyscrasia. However, PLCNA could be considered a localized plasma cell dyscrasia, in which the cutaneous clusters of plasma cells act as an extramedullary plasmacytoma that produces amyloid fibrils [5]. Though occasional cases of plasma cell monoclonality have been described, current trends consider PNCLA to be a reactive rather than a neoplastic disease [5][6][7].
PLCNA manifests as single or multiple waxy, firm, pink, yellowish, or purplish plaques or nodules (Figure 1). Atypical presentations mimicking lymphatic malformation, milia, or bullous disease have also been reported [8][9][10]. PLCNA lesions are most commonly found on the acral region, head and neck, and extremities. The plaques and nodules may have a purpuric appearance or telangiectasias visible on their surface. Although they may be asymptomatic, pain is usually present, sometimes accompanied by itching. These symptoms are thought to be due to the mass effect on adjacent tissues [11][12]. Planas-Ciudad et al. [13] reported a case of PLCNA with dystrophic calcifications on ultrasonography and subsequent histologic confirmation. Dystrophic calcifications are rarely seen in PLCNA but have been described in cases of localized amyloidosis of other organs. Patients with PLCNA are sometimes reluctant to seek treatment and do so when lesions become symptomatic or for cosmetic reasons. Spontaneous local ulceration is not uncommon, and clinical changes may sometimes occur in response to local trauma. In contrast to MA and LA, for which it is well established that repeated local trauma from scratching leads to the formation of amyloid K, there are few articles suggesting the possible role of local trauma in the deposition of the immunoglobulin light chains involved in the genesis of PLCNA [14][15].
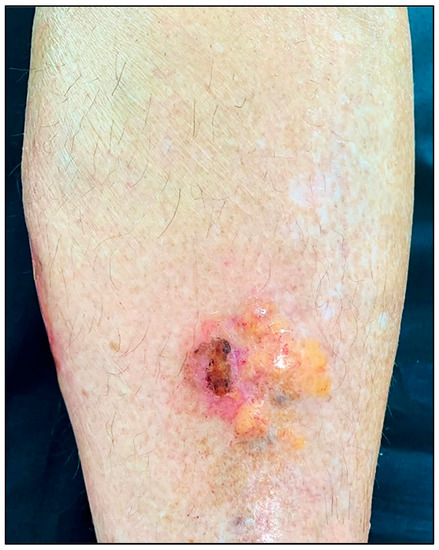
Figure 1. Patient with primary localized cutaneous nodular amyloidosis (PLCNA). Partially ulcerated yellowish nodule on the anterior aspect of the left leg.
In PLCNA dermoscopy, the most common finding appears to be a yellowish background (without structure) interrupted by whitish, scar-like lines with some hemorrhagic areas and vessels resembling fine telangiectasias. This pattern may correspond to the nodular aggregates of amyloid in the reticular dermis and subcutaneous tissue, also involving the adnexa and vessel walls with a surrounding lymphoplasmacytic infiltrate. PLCNA should be considered in the differential diagnosis of yellow-orange lesions in dermoscopy. This group comprises several histiocytic and granulomatous diseases, both inflammatory, like xanthogranuloma or sarcoidosis, and infectious, such as leishmaniasis or lupus vulgaris. In PLCNA, the unstructured yellow aggregates are different from the small yellow dots of these granulomatous diseases, which have a more pronounced inflammatory infiltrate [16].
Histopathology is considered the gold standard for diagnosing PLCNA. The nodular deposition of hyaline and eosinophilic material is usually seen in the full thickness of the dermis. Such material may also be found in small vessel walls, adnexa, and subcutaneous tissue. Variable plasma cell infiltration may occur within amyloid deposits adjacent to the vasculature. Eosinophilic material corresponding to an amyloid substance is stained with Congo red, showing a characteristic brick-red deposit. Under polarized light microscopy, an apple-green birefringence is seen. The subtype of amyloid deposits must then be determined, which reveals the κ and/or λ light chain [16]. For diagnosis, in addition to biopsies, soft-tissue imaging, preferably magnetic resonance imaging (MRI), should be requested in cases of extensive or deep involvement [17].
Systemic amyloidosis is characterized by amyloid deposition in the internal organs (mainly myocardium, liver, and kidney), which can lead to progressive loss of function and, in advanced cases, even death. Patients with systemic amyloidosis may have skin involvement, although cutaneous involvement is rare in systemic amyloidosis secondary to inflammatory disease (amyloid AA). When it does occur, it is usually mild and presents as petechiae, purpura, and/or alopecia. Primary systemic amyloidosis is caused by the proliferation of monoclonal plasma cells (amyloid AL). The underlying disorders include entities such as multiple myeloma, Waldenström’s disease, or malignant lymphomas, among others. In the primary systemic form, skin lesions are common and can vary widely. Plaques and nodules with a waxy appearance and a yellowish-orange color similar to those described in PLCNA are frequent [18]. In contrast to PLCNA, lesions are more common in mucocutaneous areas, such as the orbits and nostrils [17]. In primary systemic amyloidosis, it is frequent to find petechial, hemorrhagic, and purpuric lesions due to dermal involvement. The ulceration of papulonodular lesions is not uncommon, and when the scalp is affected, it leads to cicatricial alopecia [18]. When the dermis is extensively infiltrated by amyloid, scleroderma-like skin changes (especially on the fingers) may occur, known as ‘scleroderma amyloidosum Gottron’. Blistering and nail dystrophy have also been reported [19]. As discussed, the amyloid in both this systemic form and PLCNA is of the AL type. The histopathology of the skin lesions will show virtually identical changes in both entities [11][12].
The risk of progression of systemic amyloidosis in PLCNA was initially thought to be close to 50% based on early case reports. However, from a series of a few patients followed over time, it has now been estimated to be around 1–7%, although the rate of paraproteinemia can be as high as 40% [20][21]. Although the development of the systemic form a posteriori is uncommon, regular long-term follow-ups should be performed to rule out the progression of systemic amyloidosis. A thorough physical examination and a blood test with a complete blood count and metabolic panel are recommended at the time of diagnosis. The evaluation of the presence of a monoclonal plasma cell population outside of skin lesions is achieved by electrophoresis and immunofixation in the serum and urine, where the absence of monoclonal protein (M) indicates PLCNA. Bone marrow examination must show the absence of monoclonal plasma cells. Some authors also suggest performing initial imaging studies, such as positron emission tomography (PET) scanning, to evaluate systemic involvement. In order to rule out cardiac involvement, an electrocardiogram may be ordered [17].
In summary, amyloidosis represents a heterogeneous group of diseases with a wide variety of ethology, course, prognosis, and treatment. In particular, PLCNA can have a clinical and pathologic presentation identical to that of systemic amyloidosis. Therefore, its recognition and subsequent screenings for systemic amyloidosis are essential.
2. Primary Localized Cutaneous Nodular Amyloidosis and Sjögren’s Syndrome: A Humoral Response-Based Relationship
Sjögren’s Syndrome (SjS) is a chronic auto-immune lympho-proliferative disease characterized by the inflammation and dysfunction of the secretory glands, mainly the salivary and lacrimal glands, resulting in a dry mouth and eyes. The exocrine glands are infiltrated by T and B cells and are progressively destroyed by the cellular and humoral response. The epithelial cells of the glands are currently considered to play an important role in the pathogenesis of SjS, as they are the triggers of immune activation. Thus, epithelial cells participate in this activation by expressing ribonucleoprotein complexes (i.e., Ro/SSa and La/SSb), interacting with T cells, and producing cytokines. Although T lymphocytes play an important role in pathogenesis, B lymphocytes are the main cells involved and are also implicated in SjS-associated non-Hodgkin’s B cell lymphoma. Increased levels of cytokines are normally associated with B lymphocytes, such as IL-6 and IL-10. In addition, serum B cell activating factor (BAFF) levels are elevated and germinal center-like structures have been identified in the salivary glands of patients with SjS. Ongoing B cell hyperactivity and light chain-producing plasma cells can lead to amyloid deposits in the skin [22]. The etiopathogenesis of PLCNA in patients with SjS is summarized in Figure 2.
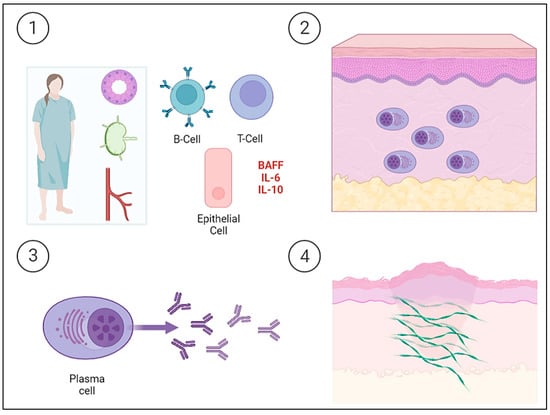
Figure 2. Schematic representation of the etiopathogenesis of Sjögren’s Syndrome (SjS) and the development of primary localized cutaneous nodular amyloidosis (PLCNA). ① The epithelial cells of the salivary gland itself and acquired immunity both play a key role in the etiopathogenesis of a patient with SjS. Epithelial cells act as initiators of the immune response by expressing ribonucleoproteins on their surface, producing cytokines, and interacting with T lymphocytes. Tissue infiltration and progressive destruction by T and B cells occur, with the latter being the fundamental cells in the etiopathogenesis and producing cytokines, such as interleukin-6 (IL-6) and interleukin-10 (IL-10). There is also an increase in serum B cell activating factor (BAFF) levels. ②, ③ B cell hyperactivity leads to the deposition of light chain-producing plasma cells in the skin. ④ The deposition of light chains leads to the formation of AL amyloid protein. The cutaneous “amyloidoma” gives rise to the characteristic plaques and nodules with a tendency to become chronic and ulcerated.
SjS should be considered in any patient with PLCNA, as it is estimated that approximately one in four patients with PLCNA also has this disease [6]. In contrast, the true prevalence of PLCNA in patients with SjS is unknown. It is thought to be very low, although it may also be an underdiagnosed entity. Besides the skin, nodular amyloidosis in SjS has been sporadically described in the lungs [23] and breasts [24]. The plasma cells in the skin produce the immunoglobulin light chain that leads to the formation of fibril and, ultimately, to the deposition of amyloid. As the clonality of plasma cells has been reported, PLCNA could, therefore, be part of the spectrum of lympho-proliferative disorders associated with SjS [25]. Interestingly, in the management of this entity, immunosuppressive agents that interfere with B cell function do not appear to be useful. Indeed, Yong et al. [26] reported an immunosuppressed renal transplant patient with IgA nephropathy who developed PLCNA despite immunosuppressive therapy with azathioprine, prednisolone, and cyclosporine. Therefore, they postulated that systemic immunosuppressants are unlikely to affect local plasma cell secretion. In addition, Tong et al. [27] reported a case wherein a patient, who also failed multiple immunosuppressive therapies, finally responded to cyclophosphamide.
Reviewing the literature to date, 26 articles were found for a total of 34 patients with PLCNA and SjS. The mean age of the patients in the reported cases was 64.47 years, with a median of 63 years (35–88). Interestingly, all but one of the cases studied were in female patients (97%). In cases where it is specified (71.33%), the median time to the diagnosis of PLCNA was 4 years (6 months–26 years), with a mean of 5.57 years. The most common location of PLCNA in patients with SjS (Figure 3) was in the lower extremities (18/34), followed by the trunk (12/34). PLCNA manifested as multifocal in distinct body areas in 20.6% of the reported cases. There is a clear preponderance of Japanese patients in those articles where ethnicity is reported. A limitation of this analysis is that the available literature is based on retrospective articles (case series and case reports).
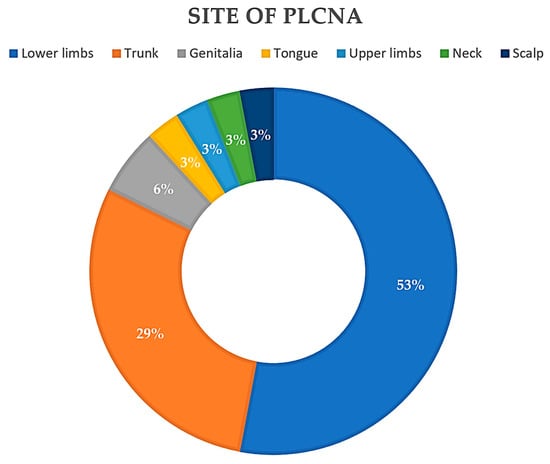
Figure 3. Pie chart. Reported locations of primary localized cutaneous nodular amyloidosis (PLCNA) in the 34 patients described to date with Sjögren Syndrome (SjS).
This entry is adapted from the peer-reviewed paper 10.3390/ijms24087378
References
- Vargas-Nevado, A.; Muñoz-Castillo, R.; López-Jiménez, P.; Bosch-García, R.; Herrera-Ceballos, E. Amiloidosis cutánea nodular primaria en paciente con síndrome de Sjögren. Piel 2015, 31, 333–335.
- Davies, K.; Collins, A.; Charlton, F.G.; Ng, W.F. Nodular localized primary cutaneous amyloidosis and primary Sjögren’s syndrome. Scand. J. Rheumatol. 2020, 49, 159–160.
- Schreml, S.; Szeimies, R.M.; Vogt, T.; Landthaler, M.; Schroeder, J.; Babilas, P. Cutaneous amyloidoses and systemic amyloidoses with cutaneous involvement. Eur. J. Dermatol. 2010, 20, 152–160.
- Cornejo, K.M.; Lagana, F.J.; Deng, A. Nodular amyloidosis derived from keratinocytes: An unusual type of primary localized cutaneous nodular amyloidosis. Am. J. Dermatopathol. 2015, 37, e129–e133.
- Hagari, Y.; Mihara, M.; Hagari, S. Nodular localized cutaneous amyloidosis: Detection of monoclonality of infiltrating plasma cells by polymerase chain reaction. Br. J. Dermatol. 1996, 135, 630–633.
- Meijer, J.M.; Schonland, S.O.; Palladini, G.; Merlini, G.; Hegenbart, U.; Ciocca, O.; Perfetti, V.; Leijsma, M.K.; Bootsma, H.; Hazenberg, B.P. Sjögren’s syndrome and localized nodular cutaneous amyloidosis: Coincidence or a distinct clinical entity? Arthritis Rheum. 2008, 58, 1992–1999.
- Grünewald, K.; Sepp, N.; Weyrer, K.; Lhotta, K.; Feichtinger, H.; Konwalinka, G.; Breathnach, S.M.; Hintner, H. Gene rearrangement studies in the diagnosis of primary systemic and nodular primary localized cutaneous amyloidosis. J. Investig. Dermatol. 1991, 97, 693–696.
- Wu, X.; Zhao, Z. Primary localized cutaneous nodular amyloidosis presenting as lymphatic malformation: A case report. Open Life Sci. 2021, 16, 781–784.
- Wang, Y.; Zhou, H.; Wang, J.; Wang, P. Primary Localized Cutaneous Nodular Amyloidosis Presenting as Milia: An Unusual Clinical Manifestation. Clin. Cosmet. Investig. Dermatol. 2022, 15, 1639–1642.
- LaChance, A.; Phelps, A.; Finch, J.; Lu, J.; Elaba, Z.; Rezuke, W.; Murphy, M.J. Nodular localized primary cutaneous amyloidosis: A bullous variant. Clin. Exp. Dermatol. 2014, 39, 344–347.
- Matsumoto, M.E.; Collins, M.K.; Raptis, A.; Jedrych, J.; Patton, T. Multifocal primary cutaneous nodular amyloidosis. Dermatol. Online J. 2017, 23, 15.
- Schwendiman, M.N.; Beachkofsky, T.M.; Wisco, O.J.; Owens, N.M.; Hodson, D.S. Primary cutaneous nodular amyloidosis: Case report and review of the literature. Cutis 2009, 84, 87–92.
- Planas-Ciudad, S.; Roé Crespo, E.; Mir-Bonafé, J.F.; Muñoz-Garza, F.Z.; Puig Sanz, L. Dystrophic Calcification in a Patient with Primary Localized Cutaneous Nodular Amyloidosis: An Uncommon Ultrasound Finding. Acta. Derm. Venereol. 2018, 98, 144–145.
- Lee, D.Y.; Kim, Y.J.; Lee, J.Y.; Kim, M.K.; Yoon, T.Y. Primary localized cutaneous nodular amyloidosis following local trauma. Ann. Dermatol. 2011, 23, 515–518.
- Caruana, D.; McCusker, S.; Harper, C.; Bilsland, D. Curious facial plaque diagnosed as nodular primary localised cutaneous amyloidosis. BMJ Case Rep. 2019, 12, e228163.
- Atzori, L.; Ferreli, C.; Matucci-Cerinic, C.; Pilloni, L.; Rongioletti, F. Primary Localized Cutaneous Nodular Amyloidosis and Limited Cutaneous Systemic Sclerosis: Additional Cases with Dermatoscopic and Histopathological Correlation of Amyloid Deposition. Dermatopathology 2021, 8, 229–235.
- Khan, N.A.J.; Nellhaus, E.; Griswold, D.; Jamil, M.O. First Case of Nodular Localized Primary Cutaneous Amyloidosis Treated with Bortezomib and Dexamethasone. J. Investig. Med. High Impact Case Rep. 2021, 9, 23247096211058488.
- Tzioufas, A.G.; Boumba, D.S.; Skopouli, F.N.; Moutsopoulos, H.M. Mixed monoclonal cryoglobulinemia and monoclonal rheumatoid factor cross-reactive idiotypes as predictive factors for the development of lymphoma in primary Sjögren’s syndrome. Arthritis Rheum. 1996, 39, 767–772.
- Fujita, Y.; Tsuji-Abe, Y.; Sato-Matsumura, K.C.; Akiyama, M.; Shimizu, H. Nail dystrophy and blisters as sole manifestations in myeloma associated amyloidosis. J. Am. Acad. Dermatol. 2006, 54, 712–714.
- Woollons, A.; Black, M.M. Nodular localized primary cutaneous amyloidosis: A long-term follow-up study. Br. J. Dermatol. 2001, 145, 105–109.
- Moon, A.O.; Calamia, K.T.; Walsh, J.S. Nodular amyloidosis: Review and long-term follow-up of 16 cases. Arch. Dermatol. 2003, 139, 1157–1159.
- Generali, E.; Costanzo, A.; Mainetti, C.; Selmi, C. Cutaneous and Mucosal Manifestations of Sjögren’s Syndrome. Clin. Rev. Allergy Immunol. 2017, 53, 357–370.
- Parambil, J.G.; Myers, J.L.; Lindell, R.M.; Matteson, E.L.; Ryu, J.H. Interstitial lung disease in primary Sjogren syndrome. Chest 2006, 130, 1489–1495.
- Kambouchner, M.; Godmer, P.; Guillevin, L.; Raphael, M.; Droz, D.; Martin, A. Low grade marginal zone B cell lymphoma of the breast associated with localised amyloidosis and corpora amylacea in a woman with long standing primary Sjogren’s syndrome. J. Clin. Pathol. 2003, 56, 74–77.
- Brown, A.J.; Spicknall, K.E.; Mutasim, D.F. Multiple lesions of primary cutaneous nodular amyloidosis in Sjögren syndrome. J. Am. Acad. Dermatol. 2012, 67, e267–e268.
- Yong, A.S.; Murphy, J.G.; Shah, N. Nodular localized cutaneous amyloidosis in an immunosuppressed patient. Int. J. Dermatol. 2015, 54, 708–709.
- Tong, P.L.; Walker, W.A.; Glancy, R.J.; Cooney, J.P.; Gebauer, K. Primary localized cutaneous nodular amyloidosis successfully treated with cyclophosphamide. Australas. J. Dermatol. 2013, 54, e12–e15.
This entry is offline, you can click here to edit this entry!