Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Virology
The human hematopoietic stem and progenitor cells (HSPCs) gave rise to a lymphoid system of human origin. The HIV research community has greatly benefitted from these hu mice. Since human immunodeficiency virus (HIV) type 1 infection results in a high-titer disseminated HIV infection, hu mice have been of great value for all types of HIV research from pathogenesis to novel therapies.
- hu mice
- HIV
- human hematopoietic stem and progenitor cells
- bNAbs
- gene therapy
- pathogenesis
1. Introduction
Human immunodeficiency virus (HIV) is a human pathogen, categorized into HIV type 1 and type 2. HIV type 1 is the virus that caused the global pandemic, while HIV type 2 is mostly confined to West Africa [1]. This review focuses on HIV type 1, unless otherwise noted. Thus, “HIV” in this review text refers to HIV-1.
Conventional small rodents used in biomedical research are resistant to HIV. Alternatives are non-human primates that are susceptible to the closely related simian immunodeficiency virus (SIV), which have been highly valuable for HIV research [2]. However, for ethical and cost reasons, work with non-human primates is limited to very few laboratories worldwide. Furthermore, data from SIV-infected non-human primates cannot be directly extrapolated to humans infected with HIV. Humanized mice were used to study HIV infection as early as the 1980s. The earliest models were the SCID-hu Thy/Liv mouse model and the hu-PBL-SCID mouse model [3] (see Box 1 for the nomenclature of humanized mice). Both models, while valuable for HIV research at the time, have significant limitations, including restriction of the analysis to the human artificial lymphoid organ under the renal capsule in the case of the SCID-hu Thy/Liv mouse model, as well as graft versus host disease (GvHD) in the case of the hu-PBL-SCID model. A new chapter in humanized mouse research began with the injection of human hematopoietic stem and progenitor cells (HSPCs) into severely immunocompromised mice. The human HSPCs engraft within three to four months and result in a hemato-lymphatic system of human origin [4]. All types of human immune cells are found in these humanized mice, which may differ in their quantity or quality according to the mouse background used. Unless otherwise indicated, hu denotes mice, e.g., hu NSG, hu BRG, humanized by transplantation of HSPCs by any method. When we use the word “humanized”, we would be referring to mice, which have been reconstituted with any type of immune cells or lymphoid tissue, including hu mice and bone marrow (B)–liver–thymus (BLT) mice.
2. What Are the Major Questions in HIV Research?
Testing of novel anti-HIV compounds and anti-HIV broadly neutralizing antibodies (bNAbs): Today, HIV doctors possess a fairly large arsenal of conventional drugs to treat HIV-infected patients [8]. Fortunately, the search for even better anti-HIV drugs continues. In particular, compounds are being investigated that target key HIV replication steps other than the conventional replication steps, e.g., compounds deactivating provirus transcription (“block-and-lock”) [9] and those that have fewer side effects or that have very long pharmacokinetic half-lives.
Furthermore, bNAbs, bi-specific antibodies, and antibody mimetics such as DARPins [10], have come to the fore in the past decade in the fight against the HIV pandemic. All of these novel agents must be tested individually or in combination, preferentially in a small-animal model, in order to obtain a pre-clinical proof of concept.
Development of cell and gene therapies to cure HIV: HIV depends on critical host factors for the completion of its replication cycle. Together with the cure of HIV infection in single patients following stem cell transplants from donors lacking CCR5 (CCR5 Δ32 homozygous donors) [11], this fact underscores the potential of genetic engineering to cure HIV. One promising approach is the genetic removal of CCR5 from hematopoietic stem and progenitor cells (HSPCs), rendering the progeny cells non-permissive to CCR5-tropic HIV strains [11]. Other approaches are based on genetically engineering either B cells for the expression of bNAbs or HIV-specific T cells resistant to HIV infection [12]. Essentially, all current clinical trials of gene therapies are based on the adoptive, either autologous or allogeneic, cell transfer of genetically engineered cells [11]. Therefore, the greatest challenge is to identify vectors for in vivo gene therapy that obviate the need for chemotherapeutic pre-conditioning [13], which is needed to provide space in adoptive cell therapies.
Exploration of experimental strategies to eradicate dormant HIV: The introduction of cART was one of the major breakthroughs in HIV medicine, resulting in a dramatic decline in morbidity and mortality among HIV-infected patients [14]. However, cART is not able to cure HIV because cART solely targets active HIV replication steps; HIV is able to hibernate in various immune cells in a latent state [15]. Various approaches have been explored to target these latently HIV-infected cells. The imminent barrier to doing so is the lack of any unequivocal marker for these cells [16].
“Shock and kill” therapies are based on the idea of inducing productive HIV replication in latently HIV-infected cells; these cells then die due to viral cytopathicity or are removed by the immune system. The “shock and kill” strategy can also be combined with the use of antibody–drug conjugates (immunotoxins) that target and kill cells that express the HIV envelope protein [17]. The neighboring non-infected cells are protected from infection by the ongoing ART [18,19]. This strategy has been repeatedly explored in clinical trials with a number of repurposed drugs [20,21]. Some of them have been shown to activate the hibernating HIV, albeit with little, if any, effect on the size of the latent reservoir [22]. There is a need for a pre-clinical in vivo model to test the wide range of possible experimental strategies.
Prophylactic or therapeutic vaccination: Since the beginning of the HIV pandemic, the HIV research community has devoted enormous effort to finding a prophylactic vaccine against HIV; these efforts have so far been in vain [23]. The major obstacles to finding a vaccine are the high mutation and recombination rates of HIV, which result in immune-escape variants. The cellular and humoral immune responses in HIV-infected individuals are certainly key elements in constraining HIV replication after acute HIV infection; however, they are deficient due to immune-escape variants. Co-evolution of the HIV envelope and antibody responses drives the emergence of bNAbs in a minority of patients. We know today that a combination of two or more bNAbs is able to suppress HIV replication in HIV-infected individuals [24]. However, the major question is the following: can we elicit bNAbs using immunogens? In the current paradigm, the priming stage is critical to the eventual induction of bNAbs. If the appropriate B-cell precursors with potential to develop into bNAbs are not stimulated at this stage, the rest of the sequential vaccine will likely fail [25,26]. In fact, in December 2022, a randomized, double-blind, placebo-controlled phase I study presented the first clinical proof-of-concept results for a germline-targeting vaccine primer [25]. Similarly, huge effort has been expended in the past few decades to develop therapeutic vaccines to enhance HIV-specific T-cell responses [27]. Some approaches have shown promising results in terms of increasing T-cell responses but ultimately have not impacted the overall goal of cART-free remission. DC-based vaccines seem to show the most promise. However, we are still a long way from a therapeutic vaccine.
Pathogenesis: The therapeutic goals listed above are inextricably linked to HIV pathogenesis. Its detailed understanding will open new avenues for HIV cure studies.
3. What Models Are Available for Biomedical HIV Research?
The value of humanized mice must be assessed in light of the alternative models for studying HIV and the question(s) being asked. The alternatives are cell lines, primary cells, and non-human primates. Each model type certainly has its place. Cell lines are optimal for studying molecular virology—by virtue of the artificial nature of cell lines, findings should then be verified in primary cells or in an in vivo setting. Primary cells have a finite lifespan and propensity to differentiate; this fact is best illustrated by the differentiation of monocytes to macrophages when adhering to the plastic of cell culture dishes. Primary cells also lack the architecture of the lymphoid organs and all associated aspects, such as cell–cell interactions, cytokine gradients, cellular diversity, etc. Notably, the readily accessible blood cells are less able to elicit ex vivo innate immune responses than cells derived from lymphoid tissue [28], pointing to the need to examine different primary cell types to obtain robust data. Furthermore, the convenience of working with cell cultures in the ambient atmosphere leads to cells being exposed to higher O2 levels than they would normally experience in vivo. Oxygen significantly affects the phenotypes of cultured cells and their transcriptional activity [29].
Chimpanzees are permissive to HIV and SIV, while other non-human primates are only permissive to SIV [30,31]. All non-human primate (NHP) models have substantial scientific value in the development of anti-HIV strategies [32]. In particular, the differential susceptibility of sooty mangabeys and rhesus macaques to SIV infection provided a unique opportunity to investigate SIV pathogenesis [2] and permitted researchers to make “dogma-changing” discoveries [2]. Notably, while SIV is closely related to HIV, they differ in various aspects [30,31]. In fact, SIV is more closely related to HIV-2 than HIV-1 [32,33]; thus, insights gained from SIV-infected NHP experiments are very welcome but should be checked for validity for HIV if possible. The main limitations to the use of NHPs are the ethical and cost aspects, which are also reflected in the rather small number of animals used in each experiment. Notably, only a few laboratories in the world have an active research program using non-human primates.
In 2004, the Manz laboratory described the generation of a human adaptive immune system in cord blood cell-transplanted mice [34]. In this seminal work, they showed that the intrahepatic injection of CD34+ human cord blood cells into newborn Rag2−/−γc−/− mice that were conditioned by irradiation, leads to the de novo development of B, T, and dendritic cells. This process also led to the formation of structured primary and secondary lymphoid organs and the production of functional immune responses, i.e., human anti-tetanus toxoid (TT) IgG antibodies following TT vaccinations and EBV-specific CD8+ T cells following EBV infection. Our group was the first to demonstrate high-titer disseminated HIV infection in these hu mice [35].
Subsequent studies using the same or a slightly modified hu mouse model have shown that HIV-infected humanized mice approximate the cardinal features of events that occur in HIV-infected humans—among others, the HIV-mediated depletion of CD4+ T cells [36,37,38,39], virus diversification [40,41,42], the establishment of a latent reservoir [36,43], and the response to anti-retroviral treatment [36]. The close approximation of HIV events to those observed in HIV-infected humans and the ease of manipulating human lymphoid cells in these models make hu mice very valuable to HIV researchers. Indeed, hu mice have enabled HIV researchers to examine a large number of questions that were previously unfeasible to investigate in an in vivo model. In fact, the HIV research community adopted this novel HIV mouse model type very quickly after the first reports.
4. What Are the Attributes of a Good HIV Mouse Model?
Ideally, a hu mouse model would have a fully fledged human-derived immune system, i.e., with all types of human immune cells, a lymphoid architecture, and the ability to mount adaptive immune responses. In addition, the mice should live for at least ten months without any symptoms whatsoever. In particular, we must pay attention to GvHD as it occurs in both hu PBL mice [44] and BLT mice [45]. GvHD mainly affects the eyes, skin, gastrointestinal tract, liver, and bone marrow [45,46]. GvHD can severely limit the lifespan of the humanized mice and, thus, the time available to conduct experiments. It can also affect the outcome of the experiment. Hu mice are less prone to GvHD.
We can distinguish three generations of hu mouse models (Figure 1). The two most prominent representatives of the first generation are the SCID-hu Thy/Liv mouse model and the hu-PBL-SCID mouse model [6]. Please, note that the SCID-hu Thy/Liv mouse model is not actually a hu mouse model as defined by this definition. Both models have substantial limitations: experiments using the SCID-hu Thy/Liv mouse model are broadly limited to the conjoint organoid, which resembles the human thymus and sustains T-cell lymphopoiesis [47]. Hu-PBL-SCID mice, with significant human blood T-lymphocyte chimerism, suffer from high levels of GvHD and mortality. The time for experimentation is very short due to GvHD [48]. Notwithstanding the limitations of hu-PBL mouse models, they are still used and have value when investigating questions that can be answered in fairly short-term experiments [49,50]. In this review, we focus primarily on a discussion of the value of second- and third-generation hu mice and BLT mice.
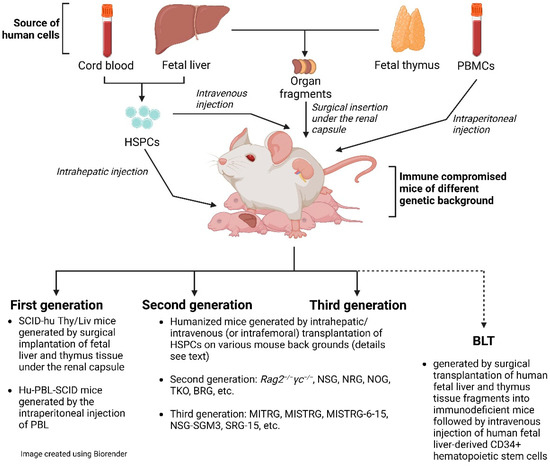
Figure 1. Cartoon of humanized mice.
The significant improvement in humanization in the second generation of hu mice is due to the very prominent immunosuppression in mice that also lack the common gamma chain (IL-2Rγ). IL-2Rγ is required for high-affinity signaling through multiple cytokine receptors, including IL-2, -4, -7, -9, -15, and -21 [51]. Notably, IL-15 is essential for NK cell development [52]. Consequently, IL-2Rγ deficiency eliminates host (murine) NK cells and improves human hematopoietic cell engraftment in immunodeficient recipient mice [4]. There are now a number of mouse strains available with the knock-out of the γ chain (γc or shortly γ), including non-obese diabetic (NOD/Scid/IL-2Rγ−/−)(NSG or NOG), NOD/Rag1−/−/IL-2Rγ−/− (NRG), and BALB/c/Rag2−/−/IL-2Rγ−/− (DKO/BRG) mice. All these mouse models have a rather high human engraftment level following transplantation with human CD34+ cells. We would like to emphasize that these second-generation hu mice still lack adequate education of human T cells on the mouse MHC molecules in the thymus and provide a suboptimal interaction between human T-cell co-receptors (CD4 and CD8) and mouse MHC [53,54]. In a very recent review by Gillgrass et al. [55], they were considered current-generation models.
We define third-generation mouse models as hu mice that, in addition to immunosuppression, express transgenes of knock-in human molecules, such as HLA, human cytokines, or murine thymic stromal lymphopoietin. In fact, hematopoiesis is a highly regulated and complex process that depends on multiple factors, such as cytokines, chemokines, adhesion factors, etc. [53]. In a human–mouse chimeric model, the interspecies cross-reactivity of key factors might be absent; thus, providing them as transgenes could improve human engraftment both quantitatively and qualitatively. With the exception of mice with human HLA-DR4 and HLA-A2 transgenes [56], which are discussed later, these third-generation hu mice also lack adequate thymic T-cell education for eventual proper T-cell function as second-generation hu mice. Thus, these alterations should result in improvements in humanization—in particular, higher engraftment levels, better functionality of immune cells, and a better lymphoid architecture. Terahara et al. refer to these hu mouse models as “next-generation mouse models” [7].
In addition to the above-described hu mice, which are based on the sole transplantation of CD34+ HSPCs, there is also the bone marrow (B)–liver–thymus (BLT) mouse model [57]. In fact, the BLT mouse model is a further development of the SCID-hu Thy/Liv described by McCune et al. in the 1980s [47,58]. The BLT mouse model is generated by first transplanting fragments of human fetal liver and thymus under the murine renal capsule, followed by irradiation and transplantation with autologous HSPCs. In fact, by providing autologous human fetal thymic fragments as a scaffold, HLA-restricted human T-cell development in vivo is supported. This model is impressive for its high engraftment level and its ability to mount a rather solid adaptive immune response [59]. However, fetal tissue is inherently limited and ethically controversial [60] and, apart from some countries, including the US and China, such fetal tissue is not available by default. BLT mice are created on various mouse backgrounds, including NSG or NRG. Particularly relevant are BLT mice constructed on the background of C57BL/6 Rag2−/−γc−/−CD47−/− mice (triple knock-out = TKO) [61]. The additional genetic inactivation of CD47 negates the requirement for CD47-signal recognition protein α (SIRP-α) signaling and induces tolerance to transplanted human HSPCs. These mice have an intact complement system and show no signs of GvHD up to 29 weeks after transplantation.
Beyond the background mouse strain, there are a large number of variables to consider in humanization, [62] such as (i) the sex of the mice, as female mice show better and faster HSPC engraftment than male mice [63]; (ii) the ultimate HSPCs and their long-term repopulation capacity [64]; (iii) the origin of the HSPCs, i.e., cord blood vs. adult vs. fetal liver; (iv) the expansion of HSPCs [65]; (v) transplantation at newborn or adult age; (vi) the type of injection of HSPCs, e.g., intrahepatic vs. intravenous injection; (vii) the number of HSPCs injected; and (viii) pre-conditioning performed with irradiation or busulfan. Hu mouse models have been customized by different laboratories according to researchers’ preferences, history, and available resources; a recent review by Stripecke et al. proposed minimum requirements for information on the humanized mouse model used when reporting on such studies [62] in order to contextualize the data presented. Beyond the more commonly used HIV mouse models, there are novel mouse models yet to be tested for HIV research, such as mouse models based on the injection of PBMCs into murine MHC class I and II double knock-out NSG [66] or NOG mice [67].
This entry is adapted from the peer-reviewed paper 10.3390/pathogens12040608
This entry is offline, you can click here to edit this entry!