Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Organic
Cyanomethylation is very useful in organic synthesis because the cyano groups can be hydrolyzed to carboxylic acids and reduced to amines, from which other functional groups can be derived. In addition, the cyano group is the structural unit of many drugs, such as piritramide, diphenozlate, and gallopamil, while acetonitrile is an ideal source of the cyanomethyl functional group due to the difficulty in breaking the C-CN bond.
- acetonitrile
- cyanomethylation
- tetrasubstituted olefins
1. Metal-Catalyzed Cyanomethylation
Transition metal catalysts have been widely used in various reactions. In 2020, Zhu’s group developed the cyanoalkylative aziridination of N-sulfonyl allylamines 1 with alkyl nitriles using Cu catalysts and 2,2’-bipyridine (Scheme 1) [28]. Many N-sulfonyl allylamine derivatives 1 and alkyl nitriles derivative were tolerated, affording the corresponding products 2 in 43–86% yields. Furthermore, enantioenriched N-tosyl-allylamines could also be utilized as substrates, giving the enantioenriched aziridines in moderate to good diastereoselectivity. Five-membered heterocycles including isoindoline and pyrrolidine 2d could be synthetized in yields of 85% and 90%, respectively.
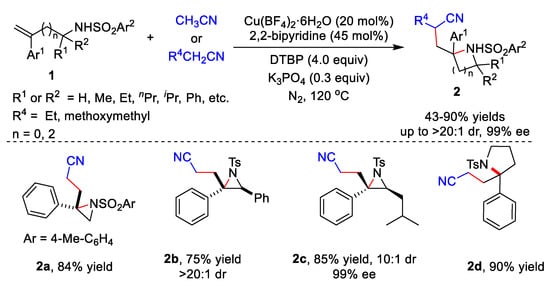
Scheme 1. Cu-catalyzed cyanoalkylative aziridination of N-sulfonyl allylamines.
In 2021, Ahmad et al. [29] reported the Cu-catalyzed cyanomethylation of various imines 3 with acetonitrile (Scheme 2). In this process, Cu(OAc)2 is used as the catalyst, and acetonitrile is used as the solvent and the CN source to synthesize arylacrylonitriles 4. The reaction conditions can tolerate a variety of substrates, including electron-donating groups, strong electron-withdrawing groups, and sterically bulky groups. However, the reaction conditions are harsh, requiring high temperatures and long reaction times. In addition, under conditions where CuCl acts as a catalyst and K2CO3 acts as a base, styrene derivatives can react with haloacetonitrile to form β,γ-unsaturated nitriles in 70–94% yields. Ahmad et al. also proposed a possible reaction mechanism (Scheme 2). First, the cyanide group of acetonitrile coordinates with copper. The complex is then attacked by (E)-N-benzylidene-4-Methylbenzenesulfonamide 3a to give intermediate 5. Elimination of methylbenzenesulfonamide 6 from 5 gives phenylacrylonitrile 4.
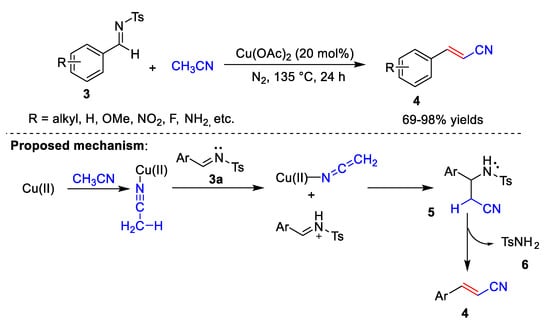
Scheme 2. Cu-catalyzed cyanomethylation.
Iron catalysts have also been used in cyanomethylation. Recently, Yao et al. [30] developed a FeCl2-catalyzed method for the cyanomethylation of amino-substituted arenes 7, using acetonitrile as the cyanomethyl source and DTBP as the oxidant (Scheme 3). Substrate adaptation studies have shown that yields are higher when substituents on the aminopyridines or anilines have an electron-withdrawing character; conversely, lower yields are obtained with electron-donating groups. In addition, the steric hindrance of the substituents greatly affects the product yields. It is noteworthy that this scheme is the amine-directed cyanomethylation reaction, and the directed amino group plays a decisive role in this reaction. When -NH2 is replaced by other groups, the cyanomethylation reaction cannot proceed. In addition, the combinations of adjacent amino and cyanomethyl groups can synthesize some valuable nitrogen-containing heterocyclic compounds, which indicates the potential practicability of this reaction. In the control experiments, when 2,2,6,6-tetramethylpiperidine-1-oxyl and butylated hydroxytoluene were added, the corresponding cyanidation products were not generated. These results indicated that a radical process is possibly involved in the reaction. Based on controlled experiments and previous literature, the authors proposed a possible reaction mechanism (Scheme 3). First, DTBP forms a tert-butoxy radical and tert-butoxy anion in the presence of iron. A proton of acetonitrile is removed by the tert-butoxy radical to form cyanomethyl radical 9. Next, the reaction can proceed via one of two ways: (a) 9 attacks the ortho-site of the amino group to afford the free radical 10, from which a proton is subsequently abstracted by the tert-butoxy anion to give 11 and t-BuOH. Finally, 11 is oxidized by Fe(III) to product 8a regenerating Fe(II). (b) FeCl2 reacts with the amino group to form 12, and 12 reacts with the cyanomethyl radical to give 13. FeCl2 is then released, and the cyanomethyl group is transferred to the aromatic ring to obtain 10. Finally, product 8a is formed through the same steps as Route (a).
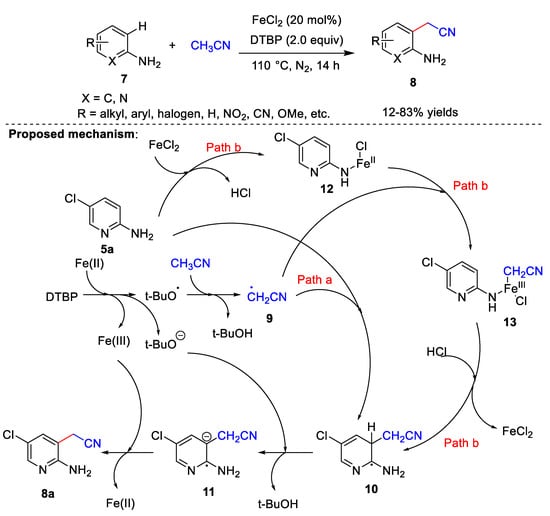
Scheme 3. Fe-catalyzed cyanomethylation.
Ni-catalyzed cyanomethylation is also important for the construction of nitrile-containing compounds. The enantioselective cyanomethylation catalyzed by Ni(II) was developed by Shibasaki and Kumagai [31,32]. Very recently, Oudeyer and co-workers established a Ni-catalyzed cyanoalkylation of ketone derivatives 14 and acetonitrile derivatives (Scheme 4) [33]. Acetonitrile was added to various isatins or activated ketones with the Ni catalyst C1, affording products 15 in up to a 99% yield. Additionally, the addition of acetonitrile derivatives to ketones required the presence of nBu4NBF4 in THF to give good product yields. Preliminary mechanistic studies showed that the Tolman-type complex, NiII-complex C1, is the key to the cyanomethylation.
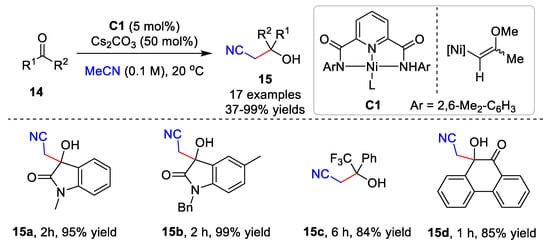
Scheme 4. Ni-catalyzed cyanomethylation.
2. Transition Metal-Free Cyanomethylation
Transition metal catalysts, which are widely used in organic synthesis, are usually expensive, pollute the environment, and add difficulties to post-treatment. In some areas, such as the pharmaceutical industry, even trace metals are not tolerated in the end product [34]. Developing environmentally friendly and economical synthetic methods is the trend of future development. As a result, metal-free catalyzed reactions have become a popular research topic in organic chemistry.
In the last few years, metal-free catalyzed direct cyanomethylation of C-H bonds, including C(sp2)-H bonds [35,36] and C(sp3)-H bonds [37], has emerged as a powerful tool in organic synthesis, which has the advantages of atom economy. In 2018, Zhang et al. [35] reported the synthesis of cyanomethylcoumarin via a cross-dehydrogenation coupling reaction between coumarin 16 and acetonitrile (Scheme 5). This reaction uses tert-butyl benzoperoxoate (TBPB) as the oxidant and potassium fluoride (KF) as the base over 16 h under a nitrogen atmosphere, with acetonitrile as both the reactant and solvent. The substrates for this methodology are broad, but require an excessive amount of solvent. Zhang et al. also proposed a reasonable mechanism for this reaction. Initially, TBPB is heated to form a benzoate radical and tert-butoxy radical. Next, cyanomethyl radical 18 is formed by abstraction of the acetonitrile proton from either the benzoate radical or the tert-butoxy radical, releasing benzoic acid or tert-butanol. Cyanomethyl radical 18 attacks coumarin 16a to give intermediate 19, which is then deprotonated to produce target product 17a.
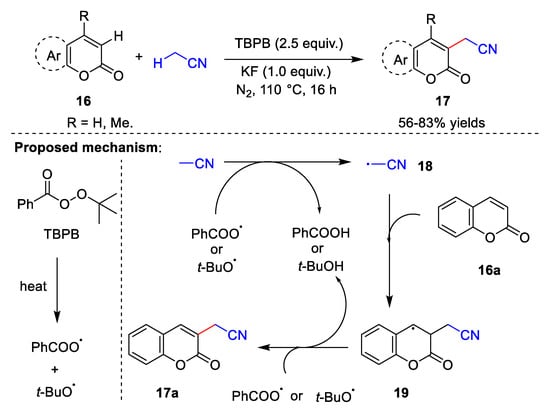
Scheme 5. Cyanomethylation with TBPB as the oxidant.
Cyanomethylation can also occur in the absence of a base. In 2021, Liu et al. [36] developed a method for the cyanomethylation of 8-aminoquinoline amides 20 (Scheme 6). This method uses only TBPB as an oxidant in the absence of metal catalysts and bases. Under these conditions, a wide range of substituents on the 8-aminoquinoline amides are tolerated, and the electronic effects and steric hindrance of the substituents have little effect on the product yields. The yields of the target products are 50–84%. It is important to note that amide’s functionality is crucial to the smooth progress of the reaction. When simple quinoline or 8-amino quinoline is used in place of 8-aminoquinoline amide, the reaction does not occur smoothly. The possible reaction mechanism proposed by Liu et al. is similar to that proposed by Zhang et al. At first, the tert-butoxy radical and benzoate radical are transferred through the thermal homolytic scission of TBPB. Subsequently, the cyanomethyl radical is generated by the hydrogen abstraction of acetonitrile by the tert-butoxy radical or benzoate radical releasing tert-butanol or benzoic acid. Thereafter, cyanomethyl radical attacks 20a to produce intermediate 22, which further reacts with the tert-butoxy radical or benzoate radical to produce required product 21a. This suggests that cyanomethylation in the absence of metal catalysis may proceed via the same reaction process.
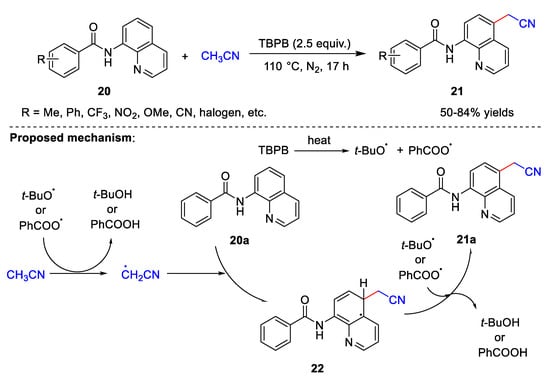
Scheme 6. Cyanomethylation with TBPB as the oxidant.
The metal-free catalyzed functionalization of unsaturated hydrocarbons involving acetonitrile is another application of cyanomethylation. Several interesting methods have been developed to provide nitrile-containing products, including the hydrofunctionalization of alkynes [38], cascade radical cyclization of alkynes [39,40], cascade radical cyclization of alkenes [41], etc. A free radical addition of acetonitrile to alkynes was developed by Liu’s group in 2019 (Scheme 7) [38]. In the presence of TBPB, alkynes 23 and acetonitrile reacted in CH3CN at 130 °C for 3 h, providing β,γ-unsaturated nitriles 24 with up to an 80% yield. The alkynes bearing aryl, alkyl, heteroaryl, cyclopropane, cyclopropane, amide, etc., units showed high compatibility with the addition reaction, affording desired products 24 in moderate to good efficiency.
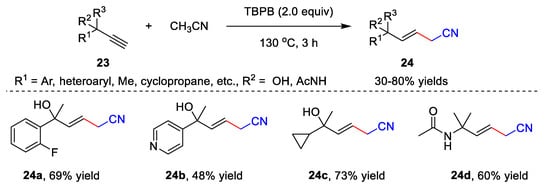
Scheme 7. Addition of acetonitrile to alkynes.
In 2020, Gao and co-workers reported an efficient cascade radical cyclization of 2-alkynylthio(seleno)anisoles 25 with acetonitrile for the construction of 3-cyanomethylated benzothio(seleno)phenes 26 (Scheme 8) [40]. Under the optimal reaction conditions, a series of desired compounds could be achieved in 45–70% yields. The author proposed a possible reaction mechanism. The DTBP first is heated to give tert-butoxy radical, which assimilates the α-H of acetonitrile to form radical I. Subsequently, generated radical I attacks the α-position of C-C triple bond of 25a to afford vinyl radical II. Finally, target product 26a is formed through a radical 5-exo-trig cyclization with the sulfur atom.
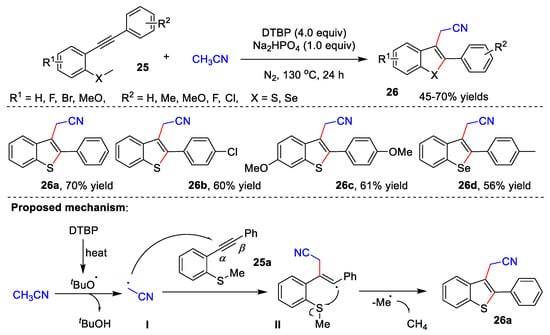
Scheme 8. Cascade radical cyclization of alkynes with acetonitrile.
In addition, Lee’s group recently reported an elegant metal-free catalyzed cyanomethylation of acetonitrile with both activated and unactivated amides 27 to afford diversified β-ketonitriles 28 with up to a 99% yield (Scheme 9) [42]. This reaction exhibits high functional group compatibility in the presence of LiHMDS. Benzamides 27 bearing not only electron-donating groups, but also electron-withdrawing groups could be utilized as substrates, giving corresponding β-ketonitriles 28 in moderate to excellent yields. Furthermore, heterocyclic acylamide, such as furan-2-carboxamide 27b and thiophene-2-carboxamide, undergo the reaction smoothly, giving the desired products in an 89% yield and 80% yield, respectively. Notably, alkyl amides 27 also display good tolerance to this method. Under the optimized conditions, benzamides 27 involving various N-substituents react well with acetonitrile and provide corresponding 28 with 25–99% yields.
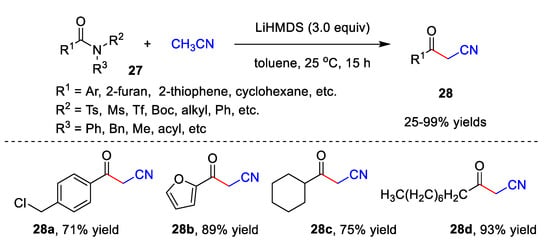
Scheme 9. Cyanomethylation of acetonitrile with amides.
This entry is adapted from the peer-reviewed paper 10.3390/catal13040761
This entry is offline, you can click here to edit this entry!