Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medical Laboratory Technology
The advancement of HIV treatment has led to increased life expectancy. People living with HIV (PLWH) are at a higher risk of developing colorectal cancers. Chronic inflammation has a key role in oncogenesis, affecting the initiation, promotion, transformation, and advancement of the disease.
- colorectal cancer
- inflammation
- metastasis
- EMT
1. Introduction
Colorectal cancer (CRC) is the third most common and second most deadly cancer worldwide. The disease affects more men than women, with a lifetime risk of 1 in 23 and 1 in 25, respectively. More younger adults (<50 years old) are seen, with an incidence of a 1.5% increase every year from 2014 to 2018 [1,2]. By, 2040, the estimated number of new CRC cases is projected to be around 3.2 million and thus poses a great burden and continuing global public health challenge [2]. Immunosuppression is the main factor that predisposes people living with human immunodeficiency virus (HIV) (PLWH) to infection-related cancers. A study by O’Neill et al. [3] found the incidence of CRC to be the same in HIV and non-HIV-infected individuals. The study included 27 studies performed in North America (18), Europe (7), the Pacific region (4), and South America (1) [3]. However, this study did not include subjects from the low-middle income countries (LMIC), particularly those from the Sub-Saharan African regions. The challenge with LMIC is the disparities in socioeconomic status. Poor living conditions expose PLWH to opportunistic infections which are exacerbated by malnutrition. Poor sanitation can lead to exposure to fecal parasites including soil-transmitted helminths [4]. These parasites can cause tissue damage to the colorectum as they migrate through the body [5]. Other factors such as microbial infections and chemical agents can also cause injury, which then triggers inflammatory responses [6,7].
Prolonged exposure to inflammatory mediators such as cytokines, chemokines, and growth factors expressed during inflammatory responses result in a chronic inflammatory state associated with the initiation of CRC [8]. This has led to the use of nonsteroidal anti-inflammatory drugs (NSAIDs) for the treatment of CRCs [9]. These drugs showed an improvement and even regression in some patients, including those diagnosed with familial adenomatous polyposis (FAP), with an inherited mutation in the tumor suppressor gene, adenomatous polyposis coli (APC) [10]. Proinflammatory cytokines such as tumor necrosis factor (TNF) and interleukin (IL)-1 trigger the activation of inflammatory signaling pathways including nuclear factor kappa B (NF-ĸB), which is needed for the transcription of cytokine genes [11]. The activation of the NF-ĸB signaling pathway also promotes CRC progression [12]. Inflammatory cytokines promote the growth of tumor cells [13]. Tumor cells release cytokines that activate the NF-ĸB pathway, pushing tumor progression towards metastatic disease [14]. The activation of the NF-ĸB signaling pathway is initiated by the toll-like receptors (TLRs). The TLRs are expressed on the surface of colonic and innate immune cells. Upon activation by a pathogenic antigen(s), a cascade of signaling molecules is stimulated to elicit immunity against the pathogen and to initiate tissue repair mechanisms [15]. Both the TLRs activation and the subsequent activation of the NF-ĸB signaling pathways are associated with oncogenesis. The TLR-2, 4, and 9 have been specifically correlated with CRC [16,17,18]. TLRs are also expressed by tumor cells [19], which prompted investigations for the use of TLRs antagonists as a treatment of choice for CRC [20] and other cancers [21]. Patients with HIV were found to be at a higher risk of developing CRC [22]. Therapeutic interventions for HIV-1 using TLRs have long been suggested [23] and have been successfully shown to eliminate HIV-infected cells in pre-clinical studies [24]. This effect on inflammatory signaling pathways links CRC and HIV, which could perhaps be a strategy for “killing two birds with one stone”. This could therefore guide the efforts to develop therapeutic strategies for preventing CRC development and progression in PLWH who are on antiretroviral therapy. Herein, we discuss key inflammatory signaling pathways, with more focus on the TLRs- NF-ĸB/IFN and associated pathways and the possibility of targeting these pathways as a therapeutic strategy that could indiscriminately benefit HIV- and non-HIV-infected CRC patients. These therapies should take into consideration the effect of parasitic or microbial infections associated with CRC development as well.
2. HIV in Colorectal Cancer
PLWH present with CRC at a younger age than their HIV-negative counterparts. The median age at CRC diagnosis was 55 years (32–73 years), and the median period of HIV infection prior to CRC diagnosis was 15 years. An HIV-infected group had more smokers or people with a previous history of smoking than the negative group. Furthermore, high levels of tumor-infiltrating lymphocytes (TILs) and a tendency of presenting with right-sided CRC was reported [25]. In concurrence with this study, HIV-infected CRC patients presented at a much younger age than those without HIV. The recurrence rate was also reported to be higher in HIV-positive (14.7%) CRC than in HIV-negative (6.8%) patients [26]. This study also supports the reports indicating that PLWH have a fourfold increased risk of developing CRC [27]. One of the mechanisms for the increased risk may be due to the Transactivator of transcription (Tat) protein, which modulates HIV-1 gene expression and facilitates the efficacy of its viral transcription. Tat is known to have oncogenic properties, and this effect was used to investigate its role in CRC oncogenesis. The main factors associated with Tat-induced CRC were its ability to significantly induce cancer cells migration, thus facilitating cancer invasiveness and metastasis. Furthermore, Tat inhibits epithelial cytodifferentiation and apoptosis, both of which are the hallmarks of cancer [27].
Conventional CRC therapy compromises immune responses and predispose patients to opportunistic infections. The use of opportunistic infection prophylaxis along with HAART was shown to decrease the risk of getting such infections [28]. Recently, HIV therapy lamivudine (3TC) was shown to attenuate CRC progression [29]. Lamivudine is a nucleoside analog and reverse transcriptase inhibitor [30]. Retrotransposon insertions [31] and repeating units [32] are associated with CRC development and progression; hence, treatment with lamivudine could halt CRC progression more profoundly in p53-mutant cell lines. The mechanism of this drug includes the induction of the interferon (IFN) response gene and the DNA damage response [29]. Xu et al. (2020) found IFNγ to play an important role in the induction of the immune checkpoint (IC) by the CRC tissue microenvironment. Apart from inducing classical ICs such as programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1), the study identified three potential novel IC-related genes induced by IFNγ. The IFNγ-inducible lysosomal thiol reductase (IFI30), guanylate binding protein1 (GBP1), and guanylate binding protein 4 (GBP4) were found to be highly expressed in CRC tumors compared to controls. The study indicated that the efficacy of IC inhibitors as cancer immunotherapy in CRC might improve by the addition of more than one IC inhibitor, with a consideration of higher instead of lower IFNγ expression levels [33].
3. Cytokine Gene Transcription Signaling in Relation to HIV-Associated CRC
Evidently, controlling HIV replication appears to play a crucial role in attenuating CRC progression [34,35]. Thus, in the efforts of finding curative HIV therapy, combination therapy with TLR agonists and broadly neutralizing antibodies (bNAbs) successfully blocked HIV-1 replication but failed to do so when used individually in people who were on anti-retroviral (ARV) therapy. Preclinical studies indicate that HIV remission could be sustained without the use of ARVs [24]. The use of HAART cannot completely eradicate HIV because of the virus’s ability to hide in the immune cells and remain dormant (HIV latency) until such time that the treatment is ceased, allowing the virus to become active and replicate. Both cytokine IL-7 and C-C chemokine ligand (CCL) 19 could increase the rate of latent HIV infection threefold. In terms of productive HIV infection, IL-7 had a fivefold increase, whilst CCL19 had a twofold increase in productive infection [36]. Latency-reversing agents (LRA) could be used to reactivate HIV transcription in dormant cells, thus stimulating the expression of surface antigens that will trigger immune responses and concurrently prevent the release of virions by HAART. However, the treatment of HIV-infected peripheral mononuclear cells (PBMCs) with TLR agonists, gene expression modulator 91 (GEM91), and a gag antisense phosphorothioate oligonucleotide showed an increased viremia caused by the induction of TLR 9 stimulation through its CpG motif. This effect was demonstrated in dose-escalation studies [37] indicating that TLR signaling contributes to challenges related to HIV treatment and possibly CRC in PLWH. Although TLRs can be beneficial in inducing immune reactivation to clear infections and prevent cancer development and progression, in certain instances, TLRs promote cancer survival and metastasis. Thus, the TLR pathway for cancer therapy is dependent on the type of TLR and the type of cell producing it. Under normal cancer immunosurveillance, TLR agonists can be beneficial to cancer cells as well. Therefore, the design of TLR-targeting agonists as well as antagonists is a promising immunotherapeutic approach to cancer [38].
TLR Signaling
Toll-like receptor family proteins are part of the innate immune system that initiates a cascade of inflammatory responses against microbial [39], parasitic [40], and viral [41] infections. There are 250 TLRs in total that have been identified thus far, with TLR 1–10 being identified in humans. The TLR family members are divided into vertebrate type (V-Type) and protostome type (P-Type). The V-Type are characterized by single-cysteine-cluster TLRs, while P-Type TLRs have several cysteine clusters. These TLRs can further be categorized into cellular (TLR 1, 2, 4, 5, 6, and 10) and intracellular membrane (TLR 3, 7, 8, and 9) TLRs (Figure 1). The TLRs are expressed on the surfaces of antigen-presenting cells (APCs) and recognize pathogen-associated molecular patterns (PAMPs) on various microorganisms. They are critical in sensing the microbiota and maintaining tolerance or triggering an immune response to pathogens. The TLRs, therefore, contribute to the prevention of gut microbial dysbiosis, associated with CRC initiation and progression [42].
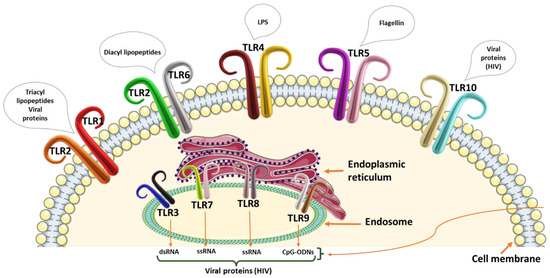
Figure 1. Representation of the human toll-like receptors on the cell membrane and the endosome with stimulatory pathogen-associated molecular patterns or danger-associated molecular patterns. Toll-like receptors 1 and 2 recognize triacyl lipopetides and viral proteins, whilst TLR2 and TLR6 recognize diacyl lipopeptides. The lipopolysaccharide (LPS) is recognized by TLR4 and flagellin by TLR5. TLR10 is specific for viral proteins including HIV protein receptors. The endosome TLR 3 recognizes double-stranded RNA (dsRNA). TLR 7 and 8 recognize single-stranded RNA (ssRNA) molecules, and TLR9 recognizes unmethylated CpG motifs. All endosome TLRs have the ability to recognize viral proteins.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15030748
This entry is offline, you can click here to edit this entry!