Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Bone is a mineralized connective tissue that is constantly renewed to maintain its mechanical properties. Osteoblasts are responsible for extracellular matrix synthesis, while osteoclasts resorb damaged bone, and the osteocytes have a regulatory role in this process, releasing growth factors and other proteins. A balanced activity among these actors is necessary for healthy bone remodeling. In obesity, several mechanisms may trigger incorrect remodeling, increasing bone resorption to the detriment of bone formation rates.
- obesity
- inflammation
- adipose tissue
- extracellular vesicles
- adipokines
- osteoblast
- osteoclast
- osteocyte
- Nc-RNAS
1. Introduction
Bone is a dynamic calcified tissue involved in physical support, visceral protection, mineral storage, hematopoiesis, and endocrine activity [28,29,30]. Bone tissue renewal is a very balanced process where hormones, cytokines, and growth factors regulate bone formation and bone resorption [31,32]. Osteoblasts are bone-forming cells derived from mesenchymal stem cells (MSCs). They produce organic extracellular matrix elements and promote their further mineralization [33,34]. On counterbalance, osteoclasts are multinucleated cells derived from the monocyte/macrophage lineage, able to resorb bone by enzymatic digestion. Bone resorption occurs predominantly by cathepsin-K and MMP-9 actions in an acidic microenvironment [35,36]. A fine balance between osteoclast and osteoblast activities controls healthy bone remodeling [37]. Additionally, osteocytes are bone cells sensitive to mechanical loads that orchestrate regulatory mechanisms to enable bone turnover. The process involves the activation of signaling routes, such as WNT, and the release of mediators, such as bone morphogenetic proteins (BMP), nitric oxide (NO), prostaglandin E2 (PGE2), sclerostatin, FGF-23 and RANKL [38,39].
Due to the systemic inflammatory condition, obesity can disrupt the balance of bone remodeling, dysregulating bone homeostasis and allowing bone loss. Two main pathways drive this condition: (i) the release of proinflammatory mediators from obese adipose tissue that reduces osteoblast activity while increasing osteoclast differentiation and resorption; and (ii) the differentiation of MSCs towards osteogenic lineage is reduced during obesity, while adipogenic differentiation is enhanced [40,41,42]. Nevertheless, other systemic alterations may also have relevance in this process, including oxidative stress, gut and microbiota physiology, vesiculated or free Nc-RNA, and hormonal changes.
2. Proinflammatory Cytokines in Bone Metabolism
Adipose tissue expansion increases pro-inflammatory cytokine expression and secretion by hypertrophic adipocytes and infiltration of M1-like macrophages and other immune cells (Figure 1). Cytokines, such as TNF-α, IL-1β, and IL-6 released by obese adipose tissue can interfere with bone cell homeostasis. Acting in isolation or in concert, they trigger intracellular signaling pathways that may lead to bone loss.
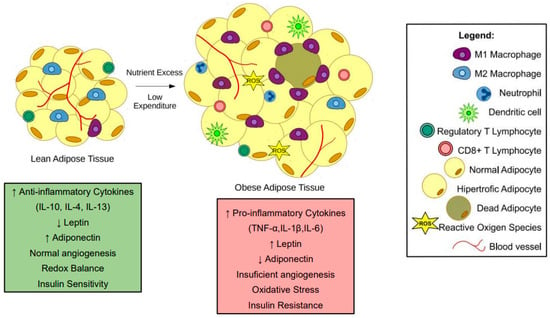
Figure 1. Changes in adipose tissue during obesity. In lean subjects, adipose tissue presents an anti-inflammatory state. Moreover, adipocytes present average triglyceride storage. During obesity, nutrient excess and low expenditure promote a hypertrophic state in adipocytes. The hypertrophic adipocytes and pro-inflammatory immune cells, such as M1-like macrophages, neutrophils, and CD8+ T cells, contribute to a low-grade inflammatory state in subjects with obesity. Most of the pro-inflammatory factors released by obese adipose tissue interfere with bone cells leading to bone loss.
TNF-α is a well know enhancer of osteoclastogenesis [43]. In vitro experiments showed that stimulation of osteoclast precursors with TNF-α and RANKL increased the number of multinucleated TRAP-positive cells [44,45]. Additionally, in vivo treatment with TNF-α increased the osteoclast numbers in mice calvaria [46]. In this context, Shinohara and collaborators (2015) demonstrated that TNF-α-induced osteoclast formation depends on double-stranded RNA-dependent protein kinase [44]. Other studies showed that TNF-α increased c-kit+/c-fms+ osteoclast precursors in high-fat diet (HFD) fed obese mice and augmented RANK expression in bone marrow macrophages [47,48]. Increased RANKL expression in TNF-α stimulated osteocytes contributed to osteoclast differentiation in vitro and in vivo [43].
In parallel, TNF-α can also modify osteoblast activity: In MC3T3-E1, osteoblasts or MSCs induced osteogenic differentiation, TNF-α decreased the expression of osteogenic transcription factors RUNX2 and Osterix, reducing the mineralization and expression of bone markers, osteocalcin (OCN), alkaline phosphatase (ALP), and bone sialoprotein (BSP) [49,50,51,52,53]. These effects are mediated by the inflammatory transcription factors ATF3 [51] and Nf-κB [53]. TNF-α upregulated miR-150-3p in osteoblasts, decreasing β-catenin expression, a key transcription factor for osteogenesis [52]. This cytokine can affect osteoblast viability, promoting apoptosis [54] and increasing MMP-9 expression [55]. TNF-β, another TNF family member, also showed deleterious impacts on bones, reducing RUNX2, Type I collagen (Col1A), OCN, and integrin β1 expression and decreasing mineralization in MSCs cultured in the osteogenic medium [56].
In vivo studies have demonstrated TNF-α-related bone loss in obese mice [57,58]. A palmitic acid-enriched HFD enhanced TNF-α serum levels in obese mice, parallel with a decrease in bone turnover markers, P1NP e CTX1 [57]. Furthermore, mice fed with a HFD presented lower trabeculae numbers and thickness and reduced trabecular bone volume, compared to lean controls. Obese TNF-α knockout mice showed less bone loss, a reduced number of femoral osteoclasts, and enhanced RUNX2 expression in MSCs, compared to HFD wildtype [58].
Increased circulating levels of IL-1β are a hallmark of the chronic, low-grade inflammation associated with obesity and related diseases [59]. IL-1β is a known inducer of osteoclast migration and resorption [60,61,62]. Combined with RANKL, IL-1β significantly increased osteoclast formation in vitro, enhancing TRAP staining and the expression of osteoclast markers, cathepsin K, OSCAR, NFATC1, Cfos, and DC-STAMP [61,62,63,64,65]. Though IL-1β increases osteoclast formation, its effect depends on the precursor subsets. Comparing the early myeloid blasts and monocytes, IL-1β increased osteoclast formation and cell diameter, mostly in myeloid blasts [61]. IL-1β can also harm other bone cells, inducing apoptosis in the MLO-Y4 osteocytic cell line [66] and MC3T3-E1 osteoblasts [67]. Osteoblasts stimulated with IL-1β exhibited a lower migration capacity [68] and released more IL-6 [69]. Indirectly, IL-1β promoted an osteoclastic supportive phenotype in osteocytes, enhancing RANKL expression [67]. Apart from triggering specific mechanisms, IL-1β also shares some signaling mechanisms with TNF-α. Lee and collaborators (2017) have shown that treatment with IL-1β and TNF-α raised CCR7 expression in both precursors and differentiated osteoclasts, increasing their migratory activity toward CCL19 and CCL21 chemokines upregulated in rheumatoid arthritis [60]. Adseverin, an actin-binding protein that regulates cell differentiation and fusion, modulates IL-1β- and TNF-α-induced osteoclastogenesis [63]. Nevertheless, Hah and collaborators (2013) showed that both IL-1β and TNF-α increased ALP activity and mineralization of periosteal osteoblasts without modifying RUNX2 and OCN expression [70].
Although osteoblasts are characterized by their activity of extracellular matrix synthesis, IL-1β enhanced metalloprotease production by osteoblasts. Ozeki and collaborators (2014) demonstrated that this cytokine potentiated MMP13 expression through ADAM28 upregulation in osteoblast-like cells [67]. Yang and collaborators (2011) have shown that IL-1β amplified the expression of MMP-9 and MMP-13 in osteoblasts [71], further contributing to bone destruction in inflammatory diseases. IL-1β may also interfere with hormonal factors produced by bone tissue, such as FGF23, which orchestrates vitamin D and phosphate serum levels [72,73]. An imbalance in FGF23 regulation, mainly due to its exceeding action, leads to a pathological mineralization process that weakens bones [74]. Corroborating in vivo studies that described greater serum levels of FGF23 in IL-1β treated mice [73], the treatment of bone slices in vitro increased hormone secretion [72]. Additionally, He and collaborators (2020) demonstrated an association between IL-1β genetic variants and osteoporosis predisposition [75]. IL-1β variants rs1143627, rs16944, and rs1143623 are related to elevated susceptibility to osteoporosis, especially in women older than 60 or with a BMI greater than 24 kg/m2 [75]. In obese mice fed a 10% corn oil-based diet, Halade and colleagues (2011) observed that bone marrow adipose tissue secreted higher concentrations of IL-1β, TNF-A, and IL-6, which increased the expression of RANK-L in stromal cells favoring osteoclast formation [76].
IL-6 is a cytokine with pleiotropic actions secreted by several cell types, including adipocytes, and its plasma levels are significantly upregulated during obesity [77]. In bone tissue, such as IL-1β and TNF-α, IL-6 indirectly stimulated osteoclastogenesis, increasing RANKL expression in osteocytes and osteoblasts [78,79,80]. Using an in vitro model of bone loss, neutralizing IL-6 antibodies had protective effects against osteoporosis, enhancing bone mineral density, trabecular number, and thickness [81]. IL-6 inhibition decreased osteoblasts’ RANKL/OPG ratio and osteoclast differentiation in a microgravity model [81]. IL-6 also inhibited osteoblast activity by downregulating the expression of the osteogenic transcription factor RUNX2 [79,81,82]. IL-6 knockout osteoblasts presented higher ALP activity and RUNX2 expression than wild-type ones. Moreover, obese IL-6 knockout mice exhibited increased trabecular bone volume, number, and thickness than HFD wild-type mice [82].
Few publications have described a combined effect of proinflammatory cytokines on bone cells. Stimulation with IL-1β, TNF-α, and IL-6 together in bone chips enhanced IL- 1β, IL-6, IL-8, TNF- α, FGF23, SOST, and OPG expression by bone cells [83]. Yokota and collaborators (2013) showed that TNF-α and IL-6 supracalvarial injections in mice raised osteoclast formation, compared to isolated injections of both cytokines [84]. Together, these data suggest the pro-osteoclastic and anti-osteoblastic effects of proinflammatory cytokines and their importance in obesity-associated bone loss (Figure 2).
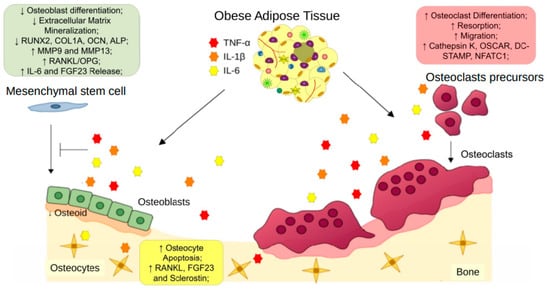
Figure 2. Obese adipose tissue-derived proinflammatory cytokines on bone remodeling. Pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, released by obese adipose tissue, exert effects on osteoclast precursors, increasing osteoclast differentiation and resorption. Moreover, these cytokines reduce the osteogenic differentiation of mesenchymal stem cells and compromise osteoblast bone formation. TNF-α, IL-6, and IL-1β also promote apoptosis and increased expression of osteoclast stimulation factors by osteocytes. The unbalanced activities between osteoblasts and osteoclasts induce osteoporosis.
This entry is adapted from the peer-reviewed paper 10.3390/cells12040521
This entry is offline, you can click here to edit this entry!