Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Intervertebral disc degeneration (IDD) and associated conditions are an important problem in modern medicine. The onset of IDD may be in childhood and adolescence in patients with a genetic predisposition. With age, IDD progresses, leading to spondylosis, spondylarthrosis, herniated disc, spinal canal stenosis. One of the leading mechanisms in the development of IDD and chronic back pain is an imbalance between pro-inflammatory and anti-inflammatory cytokines.
- cytokine
- intervertebral disc degeneration
- inflammatory
1. Introduction
Intervertebral disc degeneration (IDD) is an urgent public health problem. The main manifestation of IDD is acute and chronic back pain. It is experienced by more than 85% of people over 35 years of age [1]. Discogenic back pain is the leading cause of disability benefits in the social security system [2]. IDD is associated with damage to nearby ligaments, joints, and paravertebral muscles. Degenerated intervertebral discs (IVDs) are located lower than normal, and the apophyseal joints bear higher loads [3]. The consequence of this is osteoarthritis degeneration. The strength of the yellow ligaments decreases, which leads to their hypertrophy and protrusion of the ligaments into the spinal canal, followed by narrowing and compression of the neural structures [4].
The pathogenesis of acute and chronic pain in IDD in complex cases represents a combination of structural and mechanical deformities, as well as an increase in the activity of inflammatory mediators, including cytokines [5]. The most unfavorable in terms of the rate of progression of IDD and the formation of vertebrogenic pain syndrome is an absolute high-producing cytokine imbalance (Figure 1) [6]. This indicates that a simple measurement of one or more pro-inflammatory cytokines in the blood is not enough to make a final conclusion about the nature of changes in the cytokine balance in a patient with IDD, as well as to make an informed decision on the choice of a therapeutic strategy. for the purpose of its correction [6].
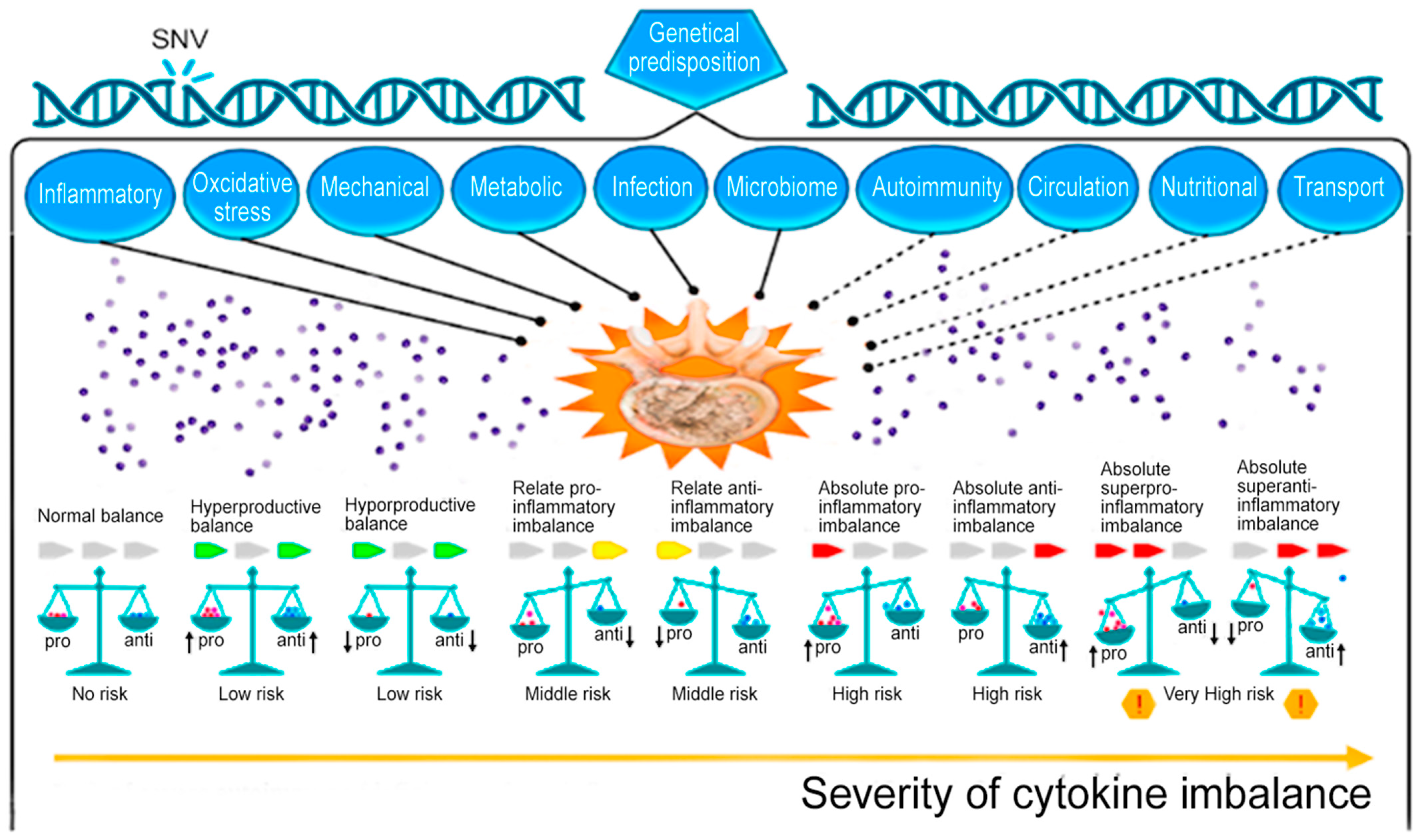
Figure 1. Potential role of normal and abnormal cytokine levels in the cytokine imbalance as a biomarker of intervertebral disc degeneration. Note: SNV—single-nucleotide variant; pro—proinflammation cytokines; anti—anti-inflammation cytokines. Note: solid line—well-studied mechanisms of IDD development; dotted line—insufficiently studied mechanisms of IDD development; up arrow—increased cytokine level; down arrow—decreased cytokine level; red exclamation point in a yellow hexagon is a dangerous situation [6].
2. General Approaches to Intervertebral Disc Degeneration Therapy
General (classical) approaches to the treatment of cytokine imbalance in patients with IDD (Figure 2) are based primarily on the symptomatic correction of inflammation and pain with systemic drugs or focal injections into the inflamed degenerating IVD or direct surgical removal of damaged IVD tissue [7]. Non-drug therapies such as exercise, weight loss, and physiotherapy are used for IDD, but only when the IDD is not severe [8]. Current treatments for IDD in sequenced or non-sequenced IVD hernias range from lifestyle changes to invasive surgical interventions, as the causes of IDD are diverse (professional habits, lifestyle, smoking, etc.) and depend on age, genetic predisposition and cellular -mediated degradation reactions accelerating after traumatic or prolonged mechanical impacts, leading to the abnormal production of cytokines and catabolic molecules by cells of degenerating IVD [9][10][11].
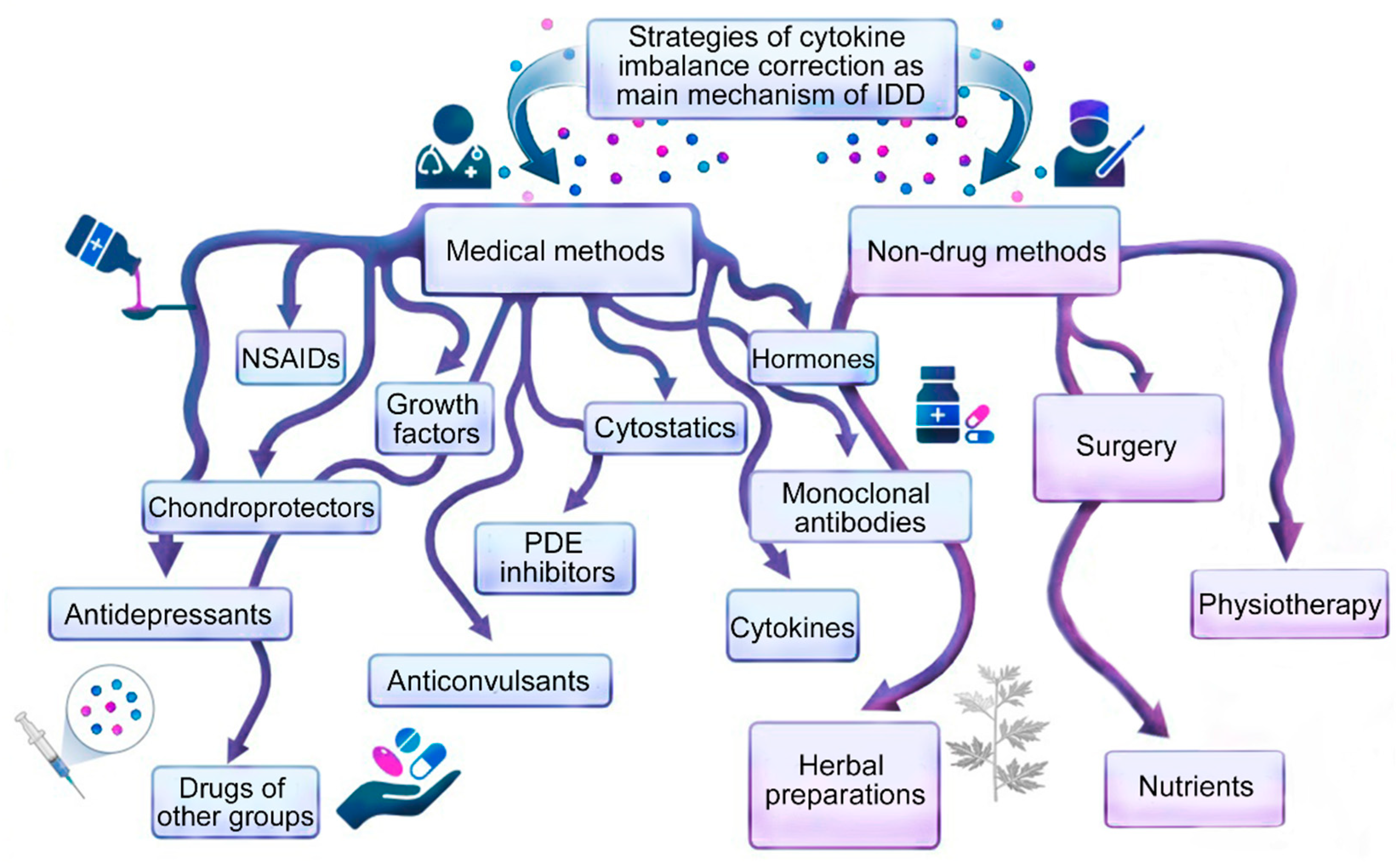
Figure 2. Methods for correcting cytokine imbalance in patients with intervertebral disc degeneration.
It is possible that an inflammatory response is involved in the onset of the disease, but it is also critical for maintaining tissue homeostasis in degenerating IVDs. In addition, optimal cytokine balance may contribute to tissue repair/regeneration of damaged IVDs [12]. Traditionally, the inflammatory response has been viewed as a negative injury mechanism that correlates with the progression of IDD. However, it remains unclear whether chronic inflammatory response in general, and cytokine imbalance in particular, is a cause or consequence of the development of IDD and herniation of IVDs. It is likely that a balanced inflammatory response is needed to reduce the rate of progression of IDD and reduce the severity of symptoms of the disease, as well as for the most complete (most possible in each specific clinical case) restoration of the function of damaged IVDs. The delicate balance between pro-inflammatory and anti-inflammatory cytokines largely determines the overall effect of the inflammatory response in patients with IDD and the expected therapeutic response to prescribed drugs. Cytokine imbalance may direct the protective immune response towards chronic inflammation (pro-inflammatory effect) or healing (anti-inflammatory effect) in patients with IDD. On the one hand, cytokine imbalance may be beneficial for IDD patients by initiating an inflammatory response in the nucleus pulposus (NP), annulus fibrosis (AF), and extracellular matrix of IVDs. On the other hand, the excessive or insufficient production of pro-inflammatory or anti-inflammatory cytokines can be harmful and initiate the development and progression of IDD [6].
The study of the molecular basis of inflammatory responses in the complex spatial and temporal organization of cellular interactions in the nucleus pulposus (NP) and annulus fibrosus (AF), as well as in the remodeling of the extracellular matrix of IVDs, can play an important role in the search for effective methods of IDD pharmacotherapy, since many of the existing therapeutic strategies do not yet achieve the expected therapeutic response [6].
3. Drugs of Other Groups
3.1. Suppressive Cytokine Anti-Inflammatory Agents
In the late 1980s, a new class of anti-inflammatory drugs was developed, called cytokine suppressive anti-inflammatory drugs (CSAIDTM). The prototype of this class of compounds is SB203580, a drug with a new mechanism of action [13]. These pyridine imidazole compounds have a potent inhibitory effect on the production of pro-inflammatory cytokines in vitro, and in vivo they can attenuate the inflammatory component of diseases in the absence of generalized immunosuppression. The molecular targets of CSAIDTM have been identified as a pair of closely related MAPK homologues, alternatively referred to as cytokine-suppressive binding protein (CSBP) [14], p38 [15], or RK [16]. p38 MAPK inhibitors have been shown to affect cytokine imbalance in a non-selective manner [17], in contrast to ERK1/2 inhibitors that block TNF-α and IL-1β, but not IL-10 [18].
3.2. Renin Inhibitors
Aliskiren ((2S,4S,5S,7S)-5-amino-N-(2-carbamoyl-2-methylpropyl)-4-hydroxy-7-{[4-methoxy-3-(3-methoxypropoxy)phenyl]methyl }-8-methyl-2-propan-2-ylnonanamide) is a renin inhibitor that reduces the level of angiotensin II with a subsequent decrease in the inflammatory response caused by the stimulation of the renin-angiotensin system [19]. Its effectiveness has been proven previously in an experimental model of osteoarthritis due to its inhibitory effect on the renin-angiotensin system present in cartilage, with a subsequent reduction in erosion [20]. Data have been accumulated on the local involvement of the renin-angiotensin system (RAS) in joint diseases [21][22]. Aliskiren has been reported to facilitate bone cell turnover in experimental rats [23]. In addition, the ability of aliskiren to reduce articular cartilage erosion was demonstrated in an experimental rat model of osteoarthritis [20]. Additionally, aliskiren is involved in the regulation of cytokine balance. Thus, the reduction of pro-inflammatory cytokines (TNF-α and IL-6) and biomarkers of oxidative stress with aliskiren was confirmed in an experiment on an animal model [24].
The relationship between the local renin-angiotensin system and the effect of RANKL on bone metabolism was revealed, which was confirmed, for example, by the study by Araújo et al. [25]. Shuai et al. [26] suggested that the local renin-angiotensin system in trabecular bone may contribute to corticoid-induced osteoporosis due to the enhanced effect of RANKL. The above effects of aliskiren may be related to the suppression of intracellular JAK-2/STAT-3 signaling, where an increase in prorenin and its receptor for STAT3 activation has been documented [27]. In addition, various studies have shown that angiotensin II stimulates the JAK-2/STAT-3 pathway [28]. Aliskiren has been shown to have a modulating effect on NOSs, including a decrease in iNOS expression and an increase in eNOS expression, which is consistent with the results of previous studies [29]. The results of these studies provide further clarification of the therapeutic effects of aliskiren against collagen-induced arthritis [30] and explain its potential role in slowing down the development of IDD. However, the efficacy and safety of using aliskiren to correct cytokine imbalance in patients with IDD needs to be clarified in the future.
3.3. Acetylcholinesterase Inhibitors
Physostigmine ((3aS-cis)-1,2,3,3a,8,8a-hexahydro-1,3a,8-trimethylpyrrolo[2,3-b]indol-5-ol methylcarbamate) is an inhibitor of acetylcholinesterase, an enzyme responsible for the breakdown of used acetylcholine. By influencing acetylcholine metabolism, physostigmine indirectly stimulates both nicotinic and muscarinic receptors due to a subsequent increase in available acetylcholine at synapses [31]. Acetylcholine appears to play a role in inflammation control as it stimulates the vagus nerve, resulting in reduced cytokine synthesis through the activation of the α7 nicotinic acetylcholine receptors. Conversely, serum acetylcholinesterase activity can have detrimental effects on bone and cartilage erosion. Cholinesterase activity has been demonstrated to increase with age, contributing to the risk and severity of arthritis in the elderly [32].
For example, the use of acetylcholinesterase inhibitors has shown a positive effect in the treatment of rheumatoid arthritis, which is associated with the control of the release of pro-inflammatory cytokines (e.g., TNF-α) and the mitigation of inflammation [33]. Ahsan et al. [34] showed that physostigmine significantly prevented the denaturation of albumin protein from hen egg and bovine serum sources in a dose-dependent manner and it also had a significant stabilizing effect on the erythrocyte membrane at higher concentrations, which may be due to the effect of removing free radicals or with the inhibition of the release of intracellular components and inflammatory mediators. In addition, physostigmine caused a significant decrease in serum levels of prostaglandin E2 and pro-inflammatory cytokine TNF-α.
3.4. Xanthines
Theophylline (1,3-dimethylxanthine) is a smooth muscle relaxant, cardiac and central nervous system stimulant. Theophylline acts as a phosphodiesterase inhibitor, adenosine receptor blocker, and histone deacetylase activator [31].
Theophylline may be a promising agent for the treatment of immunological inflammatory conditions, including the correction of cytokine imbalance in IDD. Liang et al. [35] reported an immunomodulatory effect of theophylline in an animal model. Theophylline modulates cytokine status and NO synthesis [36]. Ghasemi-Pirbaluti et al. [37] demonstrated that this drug can modulate myeloperoxidase and the production of pro-inflammatory cytokines. It has been demonstrated that theophylline has a good anti-inflammatory effect due to the activation of histone deacetylases, as well as the modulation of MMPs and other inflammatory mediators [38]. Additionally, it can modulate JAK/STAT/RANKL cytokine signaling in animals treated with complete Freund’s adjuvant, growth hormone, or a combination of the two. Thus, physostigmine plays an important role in the activation and differentiation of osteoclasts [39].
3.5. Nitrates
Nicorandil (2-(pyridine-3-carbonylamino)ethyl nitrate) is an effective oral vasodilator and antianginal agent that causes the vasodilation of arterioles and large coronary arteries by activating potassium channels [31].
It has been reported that nicorandil promotes the opening of potassium channels and has anti-inflammatory and immunomodulatory potential against inflammation in the experiment [40]. Nicorandil was found to prevent ulcerogen-induced TNF-α production [41]. The results of Gaafar et al. [39] demonstrated that this drug significantly inhibited JAK/STAT/RANKL/cytokine pathway signaling. It has immunomodulatory and anti-inflammatory effects mediated by the modulation of this signaling pathway [42]. According to Zhao et al. [43], substances that open potassium channels can protect in vitro against neuroinflammation caused by oxygen/glucose deficiency by inhibiting the activation of pro-inflammatory cytokines and the signal transduction of the Toll-like receptor 4.
3.6. Direct Oral Anticoagulants
Apixaban (1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo-piperidin-1-yl)phenyl]-4,5-dihydropyrazolo[5,4-c]pyridine-3-carboxamide) is a potent, selective, reversible direct inhibitor of Hageman factor (FXa); it is used to prevent venous thromboembolism [44]. In addition, apixaban has powerful anti-inflammatory and remodeling effects on many diseases. These effects can be explained by the inhibition of the poly-ADP ribose polymerase (PARP) enzyme on extravascular cells [45].
Previously, a significant correlation has been established between the activation of blood coagulation factors and the stimulation of various inflammatory pathways [46]. Anticoagulant therapy not only affects the coagulation cascade but can also inhibit inflammatory mediators (including pro-inflammatory cytokines), indicating a wide interaction between these processes [47]. It has been shown that FXa is an effective inducer of kinase phosphorylation, which is associated with a significant increase in the level of P-JAK2, P-STAT3, and P-MAPK in synovial tissue. Additionally, an increase in P-JAK2, P-STAT3, and P-MAPK levels was observed when Freud’s complete adjuvant group of experimental animals were stimulated with FXa. Apixaban significantly interfered with the expression of P-JAK2, P-STAT3, and P-MAPK in the synovium. The inhibition of FXa by apix-aban promoted the potent inhibition of JAK2/STAT3 and MAPK-activated pathways. It has been shown that the activation of FXa coagulation induces mRNA expression of the PAR-2 gene [48]. Bae et al. [49] reported that FXa exerts its action through the PAR-2 receptor. Consistent with the findings of Hollborn et al. [50] and Senden et al. [51], the results of El-Ghafar et al. [48] showed a significant increase in the levels of platelet growth factor and the pro-inflammatory cytokine IL-6 after FXa activation. Tissue PAR-2 levels, serum levels of platelet growth factor, and IL-6 levels were significantly reduced in all apixaban-treated rats compared to normal controls. Apixaban had a lower inhibitory effect on IL-6 compared to the other parameters listed above. This can be explained by its main action as a selective inhibitor of FXa, which subsequently leads to a significant inhibition of FXa-activated parameters such as PAR-2, platelet growth factor, and (to a lesser extent) IL-6, since the increase in the level of IL-6 expression is dependent on various signals and does not depend solely on FXa activation [52]. However, FXa-induced JAK2/STAT3 and MAPK phosphorylation may be directly related to platelet growth factor and IL-6 activation [53]. This effect may be indirectly related to PAR-2 activation, which mediates kinase and tyrosine phosphorylation, including the JAK, STAT, and MAPK pathways [54][55].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24097692
References
- Byvaltsev, V.A.; Belykh, E.G.; Stepanov, I.A.; Giers, M.; Preul, M.C. Cytokine mechanisms of degenetation of intervertebral disc. Sib. Med. J. (Irkutsk) 2015, 6. Available online: https://cyberleninka.ru/article/n/tsitokinovye-mehanizmy-degeneratsii-mezhpozvonkovogo-diska (accessed on 10 April 2023).
- Li, Y.; Samartzis, D.; Campbell, D.D.; Cherny, S.S.; Cheung, K.M.; Luk, K.D.; Karppinen, J.; Song, Y.; Cheah, K.S.; Chan, D.; et al. Two subtypes of intervertebral disc degeneration distinguished by large-scale population-based study. Spine J. 2016, 16, 1079–1089.
- Brown, S.; Rodrigues, S.; Sharp, C.; Wade, K.; Broom, N.; McCall, I.W.; Roberts, S. Staying connected: Structural integra-tion at the intervertebral disc-vertebra interface of human lumbar spines. Eur. Spine J. 2017, 26, 248–258.
- Grignon, B.; Grignon, Y.; Mainard, D.; Braun, M.; Netter, P.; Roland, J. The structure of the cartilaginous end-plates in elder people. Surg. Radiol. Anat. 2000, 22, 13–19.
- Kos, N.; Gradisnik, L.; Velnar, T. A brief review of the degenerative intervertebral disc disease. Med. Arch. 2019, 73, 421–424.
- Shnayder, N.A.; Ashhotov, A.V.; Trefilova, V.V.; Nurgaliev, Z.A.; Novitsky, M.A.; Vaiman, E.E.; Petrova, M.M.; Nasyrova, R.F. Cytokine imbalance as a biomarker of intervertebral disk degeneration. Int. J. Mol. Sci. 2023, 24, 2360.
- Kim, H.; Ham, C.H.; Kwon, W.K. Current knowledge and future therapeutic trospects in symptomatic intervertebral disc degeneration. Yonsei Med. J. 2022, 63, 199–210.
- Kloppenburg, M.; Berenbaum, F. Osteoarthritis year in review 2019: Epidemiology and therapy. Osteoarthr. Cartil. 2020, 28, 242–248.
- Okada, E.; Daimon, K.; Fujiwara, H.; Nishiwaki, Y.; Nojiri, K.; Watanabe, M.; Katoh, H.; Ishihama, H.; Fujita, N.; Tsuji, T.; et al. Ten-year longitudinal follow-up MRI study of age-related changes in thoracic intervertebral discs in asymptomatic subjects. Spine 2019, 44, E1317–E1324.
- Molinos, M.; Almeida, C.R.; Caldeira, J.; Cunha, C.; Gonçalves, R.M.; Barbosa, M.A. Inflammation in intervertebral disc degeneration and regeneration. J. R. Soc. Interface 2015, 12, 20141191.
- Weber, K.T.; Jacobsen, T.D.; Maidhof, R.; Virojanapa, J.; Overby, C.; Bloom, O.; Quraishi, S.; Levine, M.; Chahine, N.O. Developments in intervertebral disc disease research: Pathophysiology, mechanobiology, and therapeutics. Curr. Rev. Musculoskelet. Med. 2015, 8, 18–31.
- Purmessur, D.; Walter, B.; Roughley, P.; Laudier, D.; Hecht, A.; Iatridis, J. A role for TNFα in intervertebral disc degeneration: A non-recoverable catabolic shift. Biochem. Biophys. Res. Commun. 2013, 433, 151–156.
- Badger, A.M.; Bradbeer, J.N.; Votta, B.; Lee, J.C.; Adams, J.L.; Griswold, D.E. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J. Pharmacol. Exp. Ther. 1996, 279, 1453–1461.
- Lee, J.C.; Young, P.R. Role of CSB/p38/RK stress response kinase in LPS and cytokine signaling mechanisms. J. Leukoc. Biol. 1996, 59, 152–157.
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811.
- Rouse, J.; Cohen, P.; Trigon, S.; Morange, M.; Alonso-Llamazares, A.; Zamanillo, D.; Hunt, T.; Nebreda, A.R. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 1994, 78, 1027–1037.
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746.
- Foey, A.D.; Parry, S.L.; Williams, L.M.; Feldmann, M.; Foxwell, B.M.; Brennan, F.M. Regulation of monocyte IL-10 synthesis by endogenous IL-1 and TNF-alpha: Role of the p38 and p42/44 mitogen-activated protein kinases. J. Immunol. 1998, 160, 920–928.
- Choi, D.E.; Jeong, J.Y.; Lim, B.J.; Chang, Y.K.; Na, K.R.; Shin, Y.T.; Lee, K.W. Aliskiren ameliorates renal inflammation and fibrosis induced by unilateral ureteral obstruction in mice. J. Urol. 2011, 186, 694–701.
- Yan, K.; Shen, Y. Aliskiren has chondroprotective efficacy in a rat model of osteoarthritis through suppression of the local renin-angiotensin system. Mol. Med. Rep. 2017, 16, 3965–3973.
- Wang, Y.; Kou, J.; Zhang, H.; Wang, C.; Li, H.; Ren, Y.; Zhang, Y. The renin-angiotensin system in the synovium promotes periarticular osteopenia in a rat model of collagen-induced arthritis. Int. Immunopharmacol. 2018, 65, 550–558.
- Izai, M.; Miyazaki, S.; Murai, R.; Morioka, Y.; Hayashi, H.; Nishiura, M.; Miura, K. Prorenin-renin axis in synovial fluid in patients with rheumatoid arthritis and osteoarthritis. Endocrinol. Jpn. 1992, 39, 259–267.
- Goto, S.; Fujii, H.; Awata, R.; Kono, K.; Nakai, K.; Shinohara, M.; Nishi, S. Direct renin inhibitor aliskiren ameliorates low bone turnover in diabetic rat. Nephrol. Dial. Transplant. 2017, 32, iii232–iii233.
- Akpinar, E.; Halici, Z.; Cadirci, E.; Bayir, Y.; Karakus, E.; Calik, M.; Topcu, A.; Polat, B. What is the role of renin inhibition during rat septic conditions: Preventive effect of aliskiren on sepsis-induced lung injury. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 387, 969–978.
- Araújo, A.A.; Souza, T.O.; Moura, L.M.; Brito, G.A.; Aragão, K.S.; Araújo, L.S.; Medeiros, C.A.; Alves, M.S.; Araújo, R.F., Jr. Effect of telmisartan on levels of IL-1, TNF-α, down-regulated COX-2, MMP-2, MMP-9 and RANKL/RANK in an experimental periodontitis model. J. Clin. Periodontol. 2013, 40, 1104–1111.
- Shuai, B.; Yang, Y.; Shen, L.; Zhu, R.; Xu, X.; Ma, C.; Lv, L.; Zhao, J.; Rong, J. Local renin-angiotensin system is associated with bone mineral density of glucocorticoid-induced osteoporosis patients. Osteoporos. Int. 2015, 26, 1063–1071.
- Chung, S.; Kim, S.; Kim, M.; Koh, E.S.; Shin, S.J.; Park, C.W.; Chang, Y.S.; Kim, H.S. Treatment combining aliskiren with paricalcitol is effective against progressive renal tubulointerstitial fibrosis via dual blockade of intrarenal renin. PLoS ONE 2017, 12, e0181757.
- Ye, S.; Luo, W.; Khan, Z.A.; Wu, G.; Xuan, L.; Shan, P.; Lin, K.; Chen, T.; Wang, J.; Hu, X.; et al. Celastrol attenuates angiotensin II-induced cardiac remodeling by targeting STAT3. Circ. Res. 2020, 126, 1007–1023.
- Ziypak, T.; Halici, Z.; Alkan, E.; Akpinar, E.; Polat, B.; Adanur, S.; Cadirci, E.; Ferah, I.; Bayir, Y.; Karakus, E.; et al. Renoprotective effect of aliskiren on renal ischemia/reperfusion injury in rats: Electron microscopy and molecular study. Ren. Fail. 2015, 37, 343–354.
- Azouz, A.A.; Saleh, E.; Abo-Saif, A.A. Aliskiren, tadalafil, and cinnamaldehyde alleviate joint destruction biomarkers, MMP-3 and RANKL, in complete Freund’s adjuvant arthritis model: Downregulation of IL-6/JAK2/STAT3 signaling pathway. Saudi Pharm. J. 2020, 28, 1101–1111.
- Drugbank. Drugs. Available online: https://go.drugbank.com/drugs/ (accessed on 25 February 2023).
- Liu, Z.H.; Ma, Y.F.; Wu, J.S.; Gan, J.X.; Xu, S.W.; Jiang, G.Y. Effect of cholinesterase inhibitor galanthamine on circulating tumor necrosis factor alpha in rats with lipopolysaccharide-induced peritonitis. Chin. Med. J. (Engl.) 2010, 123, 1727–1730.
- van Maanen, M.A.; Vervoordeldonk, M.J.; Tak, P.P. The cholinergic anti-inflammatory pathway: Towards innovative treatment of rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 229–232.
- Ahsan, H.; Haider, I.; Mushtaq, M.N.; Qaisar, M.N.; Naqvi, F.; Asif, A. Pharmacological support to anti-arthritic prospective of physostigmine: A new approach. Inflammopharmacology 2021, 29, 1119–1129.
- Liang, B.; Jiang, S.; Zhang, Z.; Inserra, P.; Lee, J.; Solkoff, D.; Watson, R.R. Anti-inflammatory effects of theophylline: Modulation of immune functions during murine leukemia virus infection. Immunopharmacol. Immunotoxicol. 2001, 23, 307–319.
- Pal, R.; Chaudhary, M.J.; Tiwari, P.C.; Babu, S.; Pant, K.K. Protective role of theophylline and their interaction with nitric oxide (NO) in adjuvant-induced rheumatoid arthritis in rats. Int. Immunopharmacol. 2015, 29, 854–862.
- Ghasemi-Pirbaluti, M.; Motaghi, E.; Najafi, A.; Hosseini, M.J. The effect of theophylline on acetic acid induced ulcerative colitis in rats. Biomed. Pharmacother. 2017, 90, 153–159.
- Gallelli, L.; Falcone, D.; Cannataro, R.; Perri, M.; Serra, R.; Pelaia, G.; Maselli, R.; Savino, R.; Spaziano, G.; D’Agostino, B. Theophylline action on primary human bronchial epithelial cells under proinflammatory stimuli and steroidal drugs: A therapeutic rationale approach. Drug Des. Dev. Ther. 2017, 11, 265–272.
- Gaafar, A.G.A.; Messiha, B.A.S.; Abdelkafy, A.M.L. Nicorandil and theophylline can protect experimental rats against complete Freund’s adjuvant-induced rheumatoid arthritis through modulation of JAK/STAT/RANKL signaling pathway. Eur. J. Pharmacol. 2018, 822, 177–185.
- Hosseini-Tabatabaei, A.; Abdollahi, M. Potassium channel openers and improvement of toxic stress: Do they have role in the management of inflammatory bowel disease? Inflamm. Allergy Drug Targets 2008, 7, 129–135.
- El-Moselhy, M.A.; Abdel-Hamid, N.M.; Abdel-Raheim, S.R. Gastroprotective effect of nicorandil in indomethacin and alcohol-induced acute ulcers. Appl. Biochem. Biotechnol. 2009, 152, 449–459.
- Zhang, F.; Xuan, Y.; Cui, J.; Liu, X.; Shao, Z.; Yu, B. Nicorandil modulated macrophages activation and polarization via NF-κb signaling pathway. Mol. Immunol. 2017, 88, 69–78.
- Zhao, A.P.; Dong, Y.F.; Liu, W.; Gu, J.; Sun, X.L. Nicorandil inhibits inflammasome activation and Toll-like receptor-4 signal transduction to protect against oxygen-glucose deprivation-induced inflammation in BV-2 cells. CNS Neurosci. Ther. 2014, 20, 147–153.
- Barrett, Y.C.; Wang, J.; Knabb, R.; Mohan, P. Apixaban decreases coagulation activity in patients with acute deep-vein thrombosis. Thromb. Haemost. 2011, 105, 181–189.
- Schuliga, M. The inflammatory actions of coagulant and fibrinolytic proteases in disease. Mediat. Inflamm. 2015, 2015, 437695.
- Morser, J. Thrombomodulin links coagulation to inflammation and immunity. Curr. Drug Targets 2012, 13, 421–431.
- Ito, T.; Maruyama, I. Thrombomodulin: Protectorate God of the vasculature in thrombosis and inflammation. J. Thromb. Haemost. 2011, 9 (Suppl. S1), 168–173.
- El-Ghafar, O.A.M.A.; Helal, G.K.; Abo-Youssef, A.M. Apixaban exhibits anti-arthritic effects by inhibiting activated factor X-mediated JAK2/STAT3 and MAPK phosphorylation pathways. Inflammopharmacology 2020, 28, 1253–1267.
- Bae, J.S.; Yang, L.; Rezaie, A.R. Factor X/Xa elicits protective signaling responses in endothelial cells directly via PAR-2 and indirectly via endothelial protein C receptor-dependent recruitment of PAR-1. J. Biol. Chem. 2010, 285, 34803–34812.
- Hollborn, M.; Kohen, L.; Werschnik, C.; Tietz, L.; Wiedemann, P.; Bringmann, A. Activated blood coagulation factor X (FXa) induces angiogenic growth factor expression in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5930–5939.
- Senden, N.H.; Jeunhomme, T.M.; Heemskerk, J.W.; Wagenvoord, R.; van’t Veer, C.; Hemker, H.C.; Buurman, W.A. Factor Xa induces cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells. J. Immunol. 1998, 161, 4318–4324.
- Neurath, M.F.; Finotto, S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011, 22, 83–89.
- Sansone, P.; Bromberg, J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J. Clin. Oncol. 2012, 30, 1005–1014.
- Kelso, E.B.; Ferrell, W.R.; Lockhart, J.C.; Elias-Jones, I.; Hembrough, T.; Dunning, L.; Gracie, J.A.; McInnes, I.B. Expression and proinflammatory role of proteinase-activated receptor 2 in rheumatoid synovium: Ex vivo studies using a novel proteinase-activated receptor 2 antagonist. Arthritis Rheum. 2007, 56, 765–771.
- Shnayder, N.A.; Petrova, M.M.; Shesternya, P.A.; Savinova, A.V.; Bochanova, E.N.; Zimnitskaya, O.V.; Pozhilenkova, E.A.; Nasyrova, R.F. Using pharmacogenetics of direct oral anticoagulants to predict changes in their pharmacokinetics and the risk of adverse drug eactions. Biomedicines 2021, 9, 451.
This entry is offline, you can click here to edit this entry!